Von Peaks zu Genen
| Autoren |
|
| Übersetzer |
|
| Lektoren |
|
| Gutachter |
|
ÜberblickFragen:
Lernziele:
Wie benutzt man Galaxy?
Wie erhält man aus Peak‑Regionen eine Liste von Gen‑Namen?
Mit den Grundlagen von Galaxy vertraut werden
Lernen, wie man Daten aus externen Quellen bezieht
Lernen, wie man Werkzeuge ausführt
Lernen, wie Histories funktionieren
Lernen, wie man einen Workflow erstellt
Lernen, wie man seine Arbeit teilt
Geschätzte Bearbeitungszeit: 3 StundenLevel: Einsteiger IntroductoryUnterstützende Materialien:
- Datensätze
- Workflows
- galaxy-history-answer Answer Histories
- FAQs
- instances Verfügbar auf diesen Galaxy-Instanzen
Veröffentlicht: Mar 9, 2026Letzte Änderung: Mar 9, 2026Lizenz: Der Inhalt des Tutorials ist lizenziert unter der Creative Commons Attribution 4.0 International License. Das GTN Framework ist lizenziert unter MITversion Überarbeitung: 1
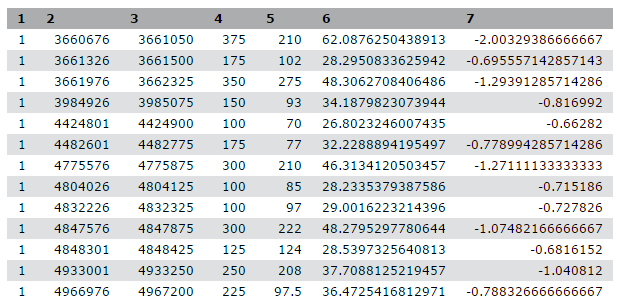
Wir sind auf eine Publikation (Li et al. 2012) mit dem Titel “The histone acetyltransferase MOF is a key regulator of the embryonic stem cell core transcriptional network “ gestoßen. Die Publikation enthält die Analyse möglicher Zielgene eines interessanten Proteins namens Mof. Die Zielgene wurden durch ChIP-seq in Mäusen gewonnen, und die Rohdaten sind über GEO verfügbar. Die Liste der Gene ist jedoch weder in den Supplements der Publikation noch in der GEO-Einreichung enthalten. Das Naheliegendste, was wir finden konnten, ist eine Datei in GEO, die eine Liste der Regionen enthält, in denen das Signal signifikant angereichert ist (so genannte Peaks):
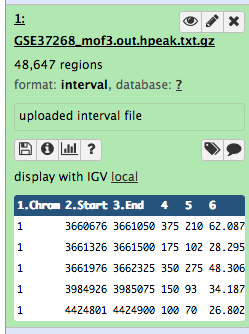
| 1 | 3660676 | 3661050 | 375 | 210 | 62.0876250438913 | -2.00329386666667 |
| 1 | 3661326 | 3661500 | 175 | 102 | 28.2950833625942 | -0.695557142857143 |
| 1 | 3661976 | 3662325 | 350 | 275 | 48.3062708406486 | -1.29391285714286 |
| 1 | 3984926 | 3985075 | 150 | 93 | 34.1879823073944 | -0.816992 |
| 1 | 4424801 | 4424900 | 100 | 70 | 26.8023246007435 | -0.66282 |
Tabelle 1 Teilprobe der verfügbaren Datei
Das Ziel dieser Übung ist es, die Liste der genomischen Regionen in eine Liste möglicher Zielgene umzuwandeln.
Kommentar: Ergebnisse können variierenIhre Ergebnisse können sich aufgrund unterschiedlicher Versionen von Tools, Referenzdaten, externen Datenbanken oder aufgrund stochastischer Prozesse in den Algorithmen geringfügig von den in diesem Tutorium vorgestellten unterscheiden.
AgendaIn diesem Tutorium werden wir uns mit folgenden Themen beschäftigen:
Vorbehandlungen
Praktische Übung: Open Galaxy
- Navigieren Sie zu einer Galaxy-Instanz: die von Ihrem Ausbilder empfohlene oder eine aus der Liste Galaxy-Instanz im Kopf dieser Seite
Anmelden oder registrieren (oberes Feld)
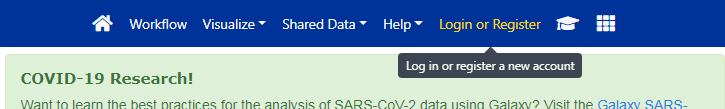
Die Galaxy-Schnittstelle besteht aus drei Hauptteilen. Auf der linken Seite sind die verfügbaren Werkzeuge aufgelistet, auf der rechten Seite wird Ihr Analyseverlauf aufgezeichnet, und im mittleren Bereich werden die Werkzeuge und Datensätze angezeigt.
 Open image in new tab
Open image in new tabBeginnen wir mit einer neuen History.
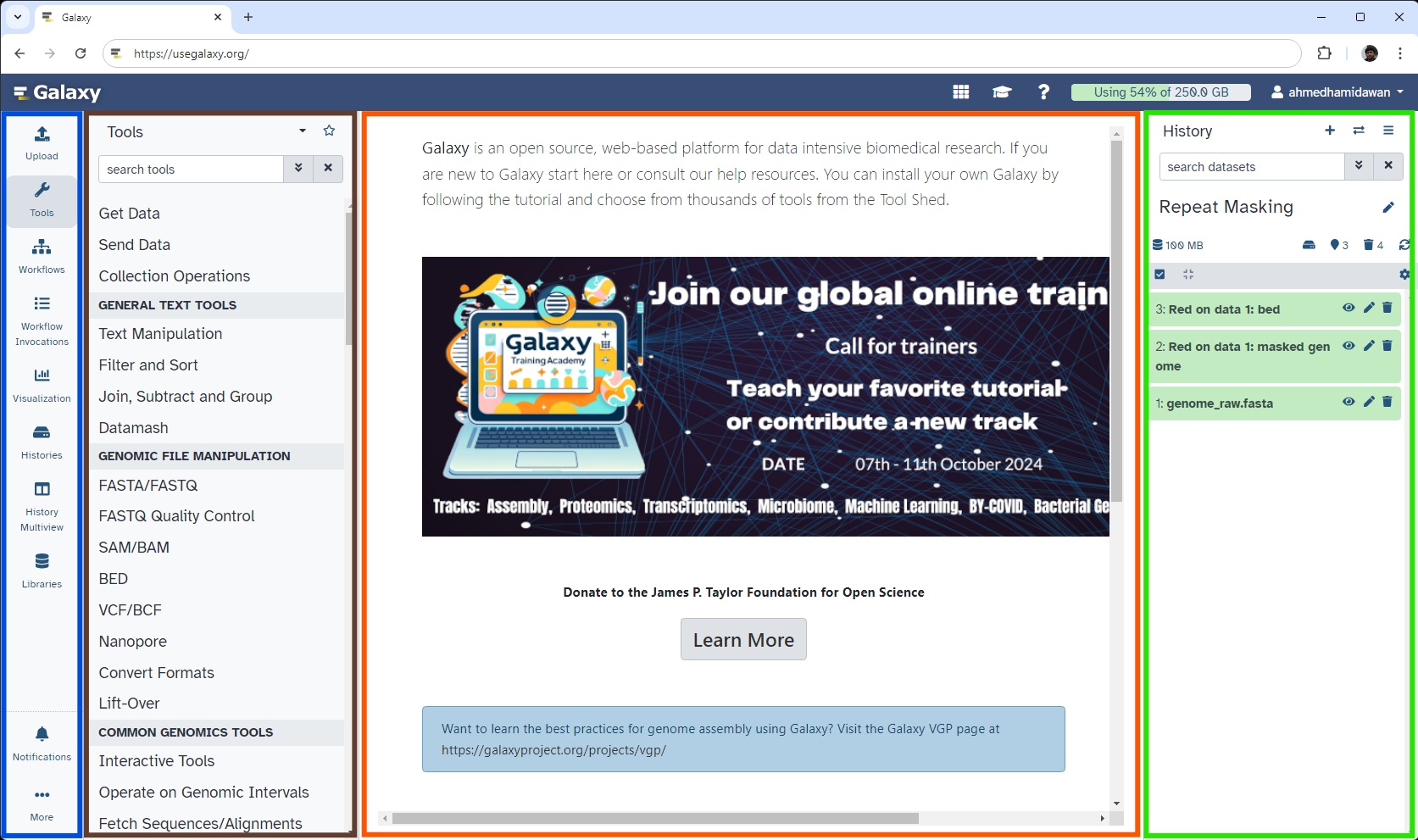
Praktische Übung: Historie erstellen
Stellen Sie sicher, dass Sie einen leeren Analyseverlauf haben.
Um einen neuen Verlauf zu erstellen, klicken Sie einfach auf das Symbol new-history am oberen Rand des Verlaufsfensters:
Benennen Sie Ihren Verlauf, damit Sie ihn leicht erkennen können
Klicken Sie auf den Titel des Verlaufs (standardmäßig ist der Titel
Unnamed history)
- Geben Sie als Namen
Galaxy Introductionein- Drücken Sie Eingabe

Daten-Upload
Praktische Übung: Daten-Upload
- Laden Sie die Liste der Peak-Regionen (die Datei
GSE37268_mof3.out.hpeak.txt.gz) von GEO auf Ihren Computer herunterKlicken Sie auf die Schaltfläche “Hochladen” in der oberen linken Ecke der Benutzeroberfläche
- Drücken Sie Lokale Dateien auswählen und suchen Sie die Datei auf Ihrem Computer
- Select
intervalas Type- Drücken Sie Start
- Drücken Sie Schließen
Warten Sie, bis der Upload abgeschlossen ist. Galaxy wird die Datei automatisch entpacken.
Danach sehen Sie Ihren ersten Eintrag in der Historie im rechten Fensterbereich von Galaxy. Es durchläuft den grauen (Vorbereitung/Warteschlange) und gelben (läuft) Status und wird schließlich grün (Erfolg):
Das direkte Hochladen von Dateien ist nicht die einzige Möglichkeit, Daten in Galaxy zu erhalten
- Kopieren der Linkposition
Klicken Sie auf galaxy-upload Daten hochladen am oberen Rand der Werkzeugleiste
- Wählen Sie galaxy-wf-edit Daten einfügen/holen
Fügen Sie den/die Link(s) in das Textfeld ein
Ändern Sie Type (set all): von “Auto-detect” auf
intervalDrücken Sie Start
- Schließen Sie das Fenster
Es gibt weitere Optionen für fortgeschrittene Benutzer.
Kommentar: Intervall-DateiformatDas Intervall-Format ist ein Galaxy-Format zur Darstellung genomischer Intervalle. Es ist tabulatorgetrennt, hat aber die zusätzliche Anforderung, dass drei der Spalten die folgenden Spezifikationen beinhalten müssen:
- Chromosomen-ID
- Startposition (0-basiert)
- Endposition (end-exclusive)
Eine optionale Strangspalte kann ebenfalls angegeben werden, und eine anfängliche Kopfzeile kann verwendet werden, um die Spalten zu beschriften, die nicht in einer bestimmten Reihenfolge angeordnet sein müssen. Im Gegensatz zum BED-Format (siehe unten) können auch beliebige zusätzliche Spalten vorhanden sein.
Weitere Informationen über Formate, die in Galaxy verwendet werden können, finden Sie auf der Galaxy Data Formats page.
Praktische Übung: Attribute einer Datei untersuchen und bearbeiten
Klicken Sie auf die Datei im Verlaufsfenster
Einige Metainformationen (z.B. Format, Referenzdatenbank) über die Datei und die Kopfzeile der Datei werden dann zusammen mit der Anzahl der Zeilen in der Datei (48.647) angezeigt:
Klicken Sie auf das galaxy-eye (Auge) Symbol (Daten ansehen) in Ihrem Datensatz in der Historie
Der Inhalt der Datei wird im zentralen Feld angezeigt
Klicken Sie auf das galaxy-pencil (Bleistift)-Symbol (Attribute bearbeiten) in Ihrem Datensatz in der Historie
Ein Formular zur Bearbeitung von Datensatzattributen wird im zentralen Bereich angezeigt
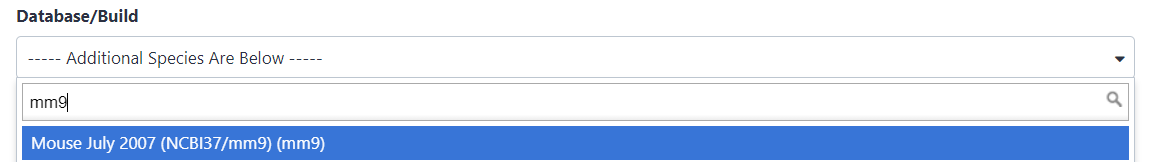
Suche nach
mm9im Datenbank/Build-Attribut und Auswahl vonMouse July 2007 (NCBI37/mm9)(die Publikaton sagt uns, dass die Peaks vonmm9stammen)
- Klicken Sie auf Speichern am oberen Rand
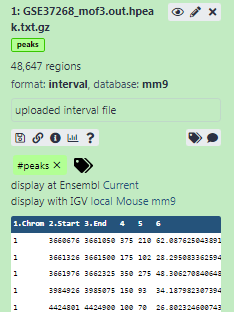
Hinzufügen eines Tags namens
#peakszum Datensatz, um ihn in der Historie besser verfolgen zu könnenDatensätze können getaggt werden. Dies vereinfacht die Verfolgung von Datensätzen über die Galaxy-Schnittstelle. Tags können eine beliebige Kombination von Buchstaben oder Zahlen enthalten, dürfen aber keine Leerzeichen enthalten.
Um einen Datensatz zu taggen:
- Klicken Sie auf den Datensatz, um ihn zu erweitern
- Klicken Sie auf Add Tags galaxy-tags
- Fügen Sie tag text hinzu. Tags, die mit
#beginnen, werden automatisch an die Ausgaben von Tools weitergegeben, die diesen Datensatz verwenden (siehe unten).- Drücken Sie Enter
- Stellen Sie sicher, dass das Tag unter dem Namen des Datensatzes erscheint
Tags, die mit
#beginnen, sind speziell!Sie werden Name-Tags genannt. Das Besondere an diesen Tags ist, dass sie sich vererben: Wenn ein Datensatz mit einem Name-Tag gekennzeichnet ist, erben alle Ableitungen (Kinder) dieses Datensatzes automatisch dieses Tag (siehe unten). Die folgende Abbildung erklärt, warum dies so nützlich ist. Betrachten Sie die folgende Analyse (die Zahlen in Klammern entsprechen den Datensatznummern in der Abbildung unten):
- Ein Satz von Vorwärts- und Rückwärts-Reads (Datensätze 1 und 2) wird mit Bowtie2 gegen eine Referenz gemappt, wodurch Datensatz 3 erzeugt wird;
- Datensatz 3 wird verwendet, um die Read Coverage mit BedTools Genome Coverage getrennt für
+und-Stränge. Dies erzeugt zwei Datensätze (4 und 5 für plus bzw. minus);- Die Datensätze 4 und 5 werden als Input für die Macs2 broadCall -Datensätze verwendet, die die Datensätze 6 und 8 erzeugen;
- Die Datensätze 6 und 8 werden mit BedTools Intersect mit den Koordinaten von Genen (Datensatz 9) geschnitten, wodurch die Datensätze 10 und 11 entstehen.
Betrachten wir nun, dass diese Analyse ohne Namens-Tags durchgeführt wird. Dies ist auf der linken Seite der Abbildung zu sehen. Es ist schwer nachzuvollziehen, welche Datensätze “Plus”-Daten und welche “Minus”-Daten enthalten. Enthält zum Beispiel Datensatz 10 “Plus”-Daten oder “Minus”-Daten? Wahrscheinlich “minus”, aber sind Sie sicher? Bei einem kleinen Verlauf wie dem hier gezeigten ist es möglich, dies manuell nachzuvollziehen, aber wenn der Umfang eines Verlaufs wächst, wird es sehr schwierig.
Die rechte Seite der Abbildung zeigt genau die gleiche Analyse, jedoch unter Verwendung von Namens-Tags. Als die Analyse durchgeführt wurde, waren die Datensätze 4 und 5 mit
#plusbzw.#minusgekennzeichnet. Als sie als Eingaben in Macs2 verwendet wurden, übernahmen die Datensätze 6 und 8 automatisch diese Markierungen und so weiter… Daher ist es einfach, beide Zweige (plus und minus) dieser Analyse zu verfolgen.Weitere Informationen finden Sie in einem dedizierten #nametag-Tutorial.
Der Datensatz sollte nun in der Historie wie folgt aussehen

Um die mit diesen Peak-Regionen verwandten Gene zu finden, benötigen wir auch eine Liste von Genen in Mäusen, die wir von UCSC erhalten können.
Praktische Übung: Daten-Upload von UCSC
Suche nach
UCSC Mainin der Werkzeug-Suchleiste (oben links)
Klicken Sie auf
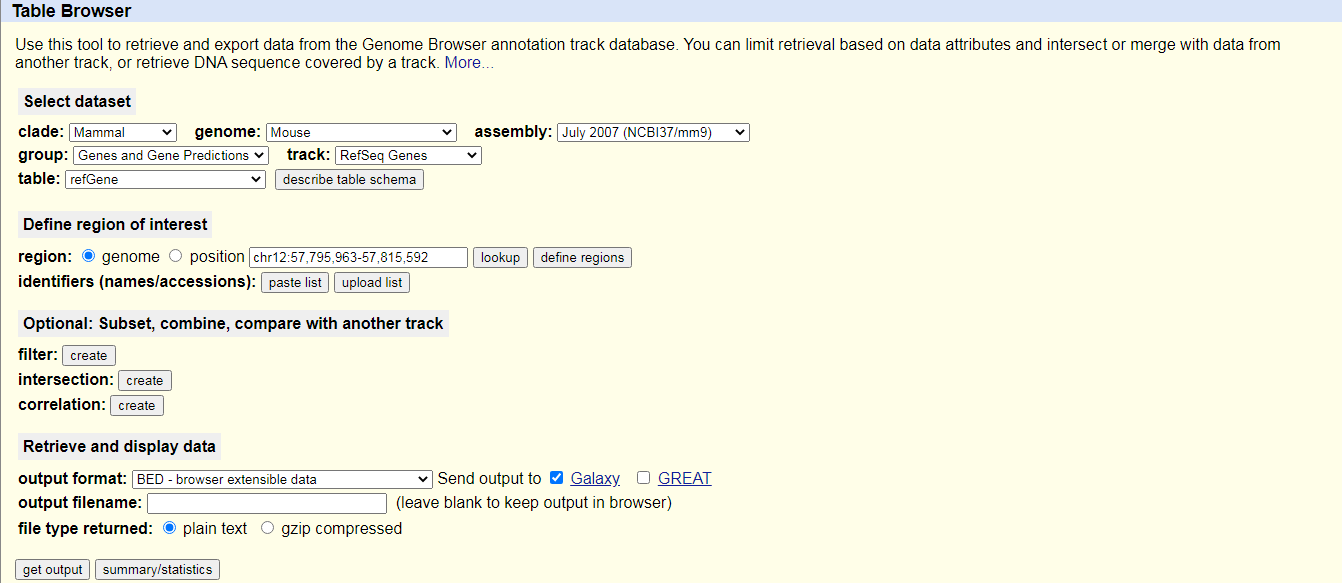
UCSC MaintoolSie werden zum UCSC-Tabellenbrowser weitergeleitet, der in etwa wie folgt aussieht:
- Setzen Sie die folgenden Optionen:
- “Klade “:
Mammal- “Genom “:
Mouse- “Assembly”:
July 2007 (NCBI37/mm9)- “Gruppe “:
Genes and Gene Predictions- “Spur “:
RefSeq Genes- “Tabelle “:
refGene- “Region “:
genome- “Ausgabeformat “:
BED - browser extensible data- “Ausgabe senden an “:
Galaxy(nur)Klicken Sie auf die Schaltfläche Ausgabe erhalten
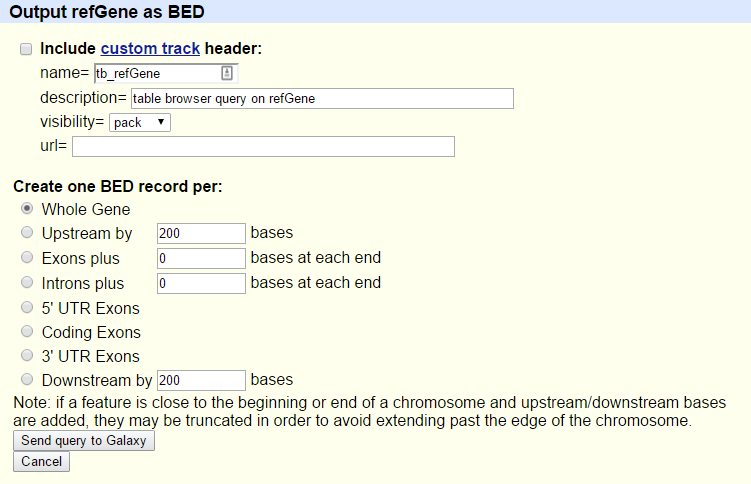
Sie sehen den nächsten Bildschirm:
- Stellen Sie sicher, dass “Create one BED record per “ auf
Whole Genegesetzt ist- Klicken Sie auf die Schaltfläche Abfrage an Galaxy senden
- Warten, bis der Upload beendet ist
Benennen Sie unseren Datensatz in etwas Wiedererkennbareres wie
Genesum
- Klicken Sie auf das galaxy-pencil Bleistift-Symbol für den Datensatz, um seine Attribute zu bearbeiten
- Ändern Sie im zentralen Bereich das Feld Name in
Genes- Klicken Sie auf die Schaltfläche Speichern
- Hinzufügen eines Tags namens
#geneszum Datensatz, um ihn in der Historie besser verfolgen zu können
Kommentar: BED-DateiformatDas BED - Browser Extensible Data-Format bietet eine flexible Möglichkeit zur Kodierung von Genregionen. BED-Zeilen haben drei Pflichtfelder:
- Chromosomen-ID
- Startposition (0-basiert)
- Endposition (end-exclusive)
Es kann bis zu neun zusätzliche optionale Felder geben, aber die Anzahl der Felder pro Zeile muss in jedem einzelnen Datensatz konsistent sein.
Weitere Informationen dazu finden Sie unter UCSC, einschließlich einer Beschreibung der optionalen Felder.
Jetzt haben wir alle Daten gesammelt, die wir für unsere Analyse benötigen.
Teil 1: Naiver Ansatz
Zunächst wird mit einem “naiven” Ansatz versucht, die Gene zu identifizieren, mit denen die Peak-Regionen verbunden sind. Wir werden Gene identifizieren, die sich mindestens 1bp mit den Peakregionen überschneiden.
Dateivorbereitung
Werfen wir einen Blick auf unsere Dateien, um zu sehen, was wir hier haben.
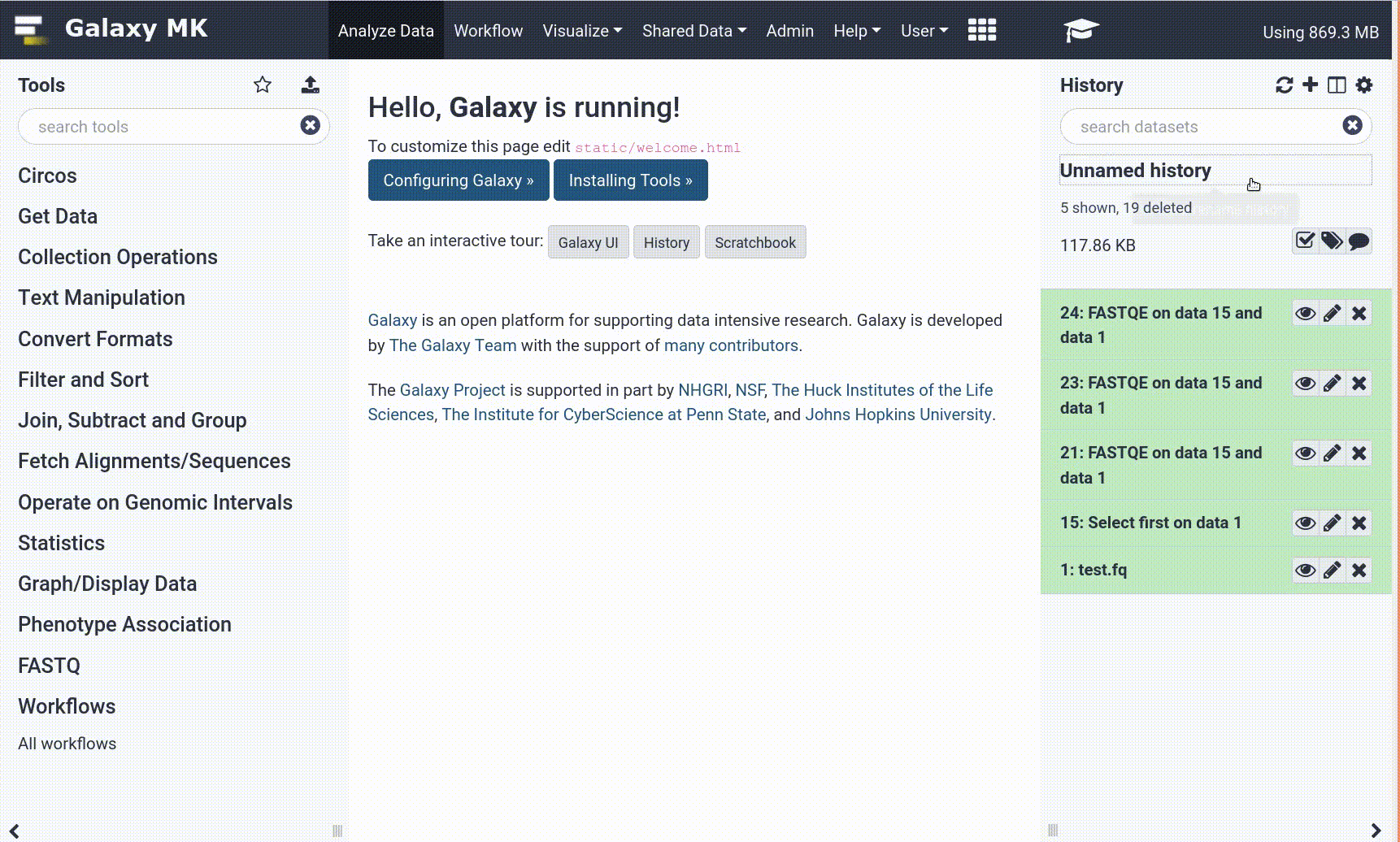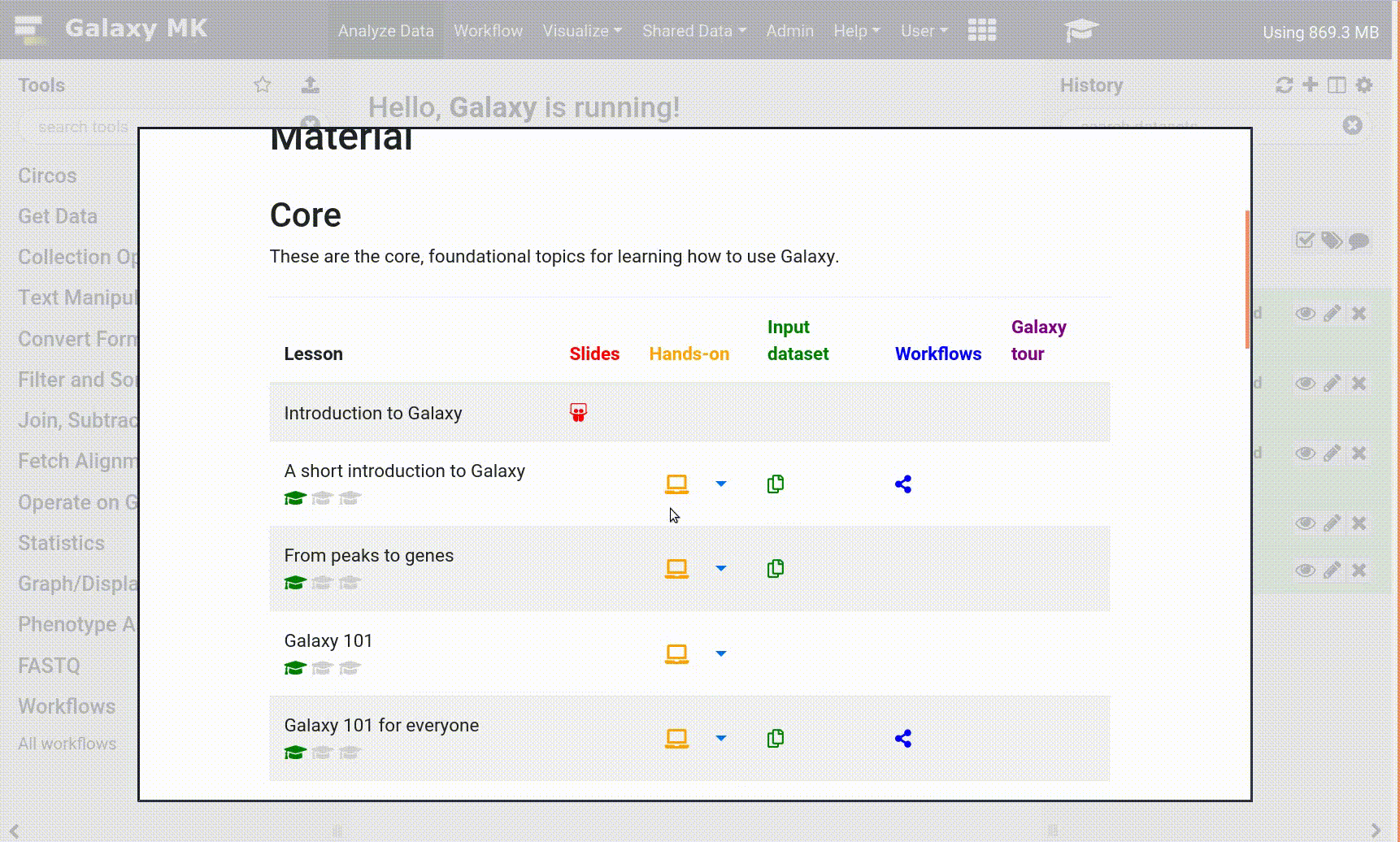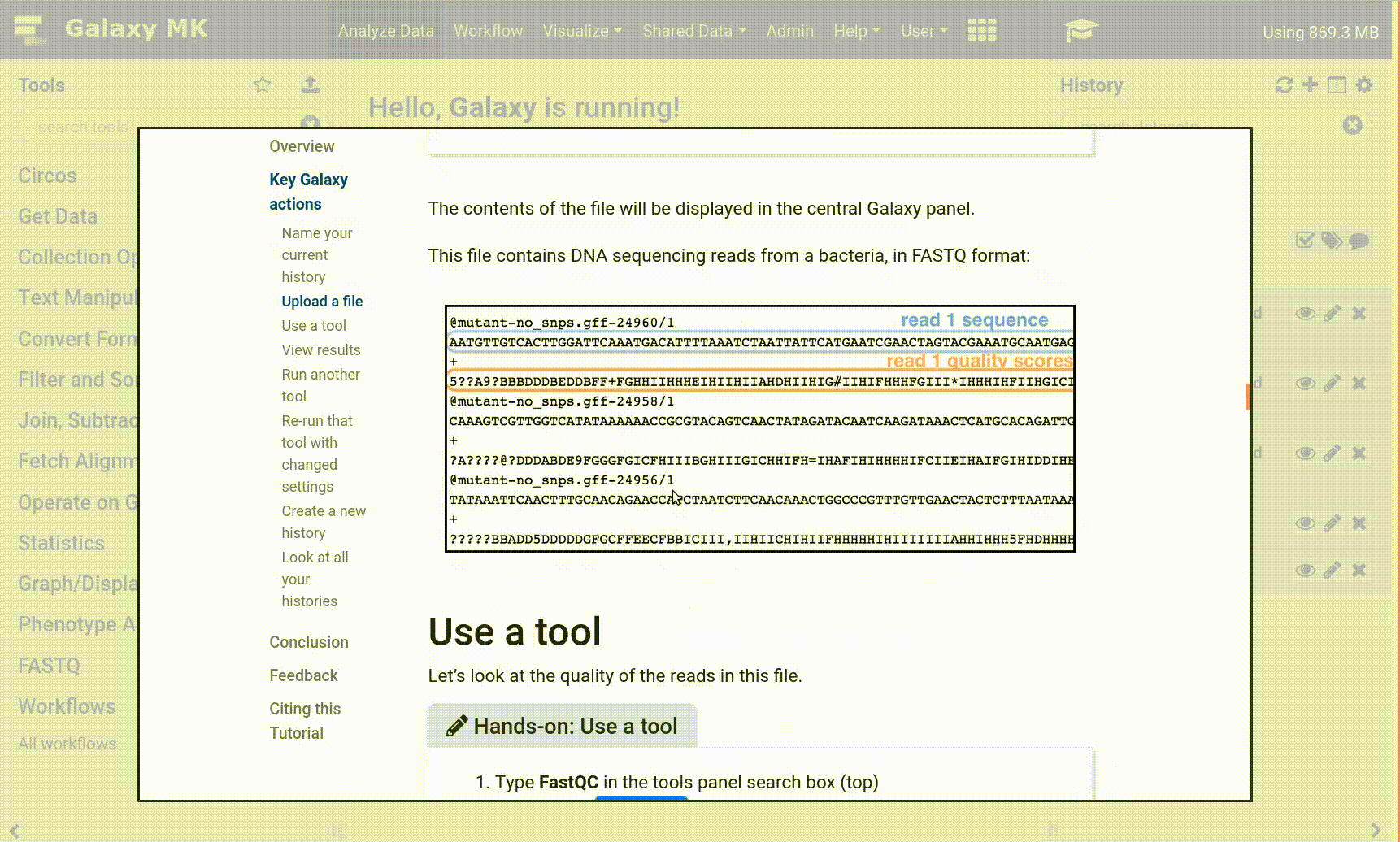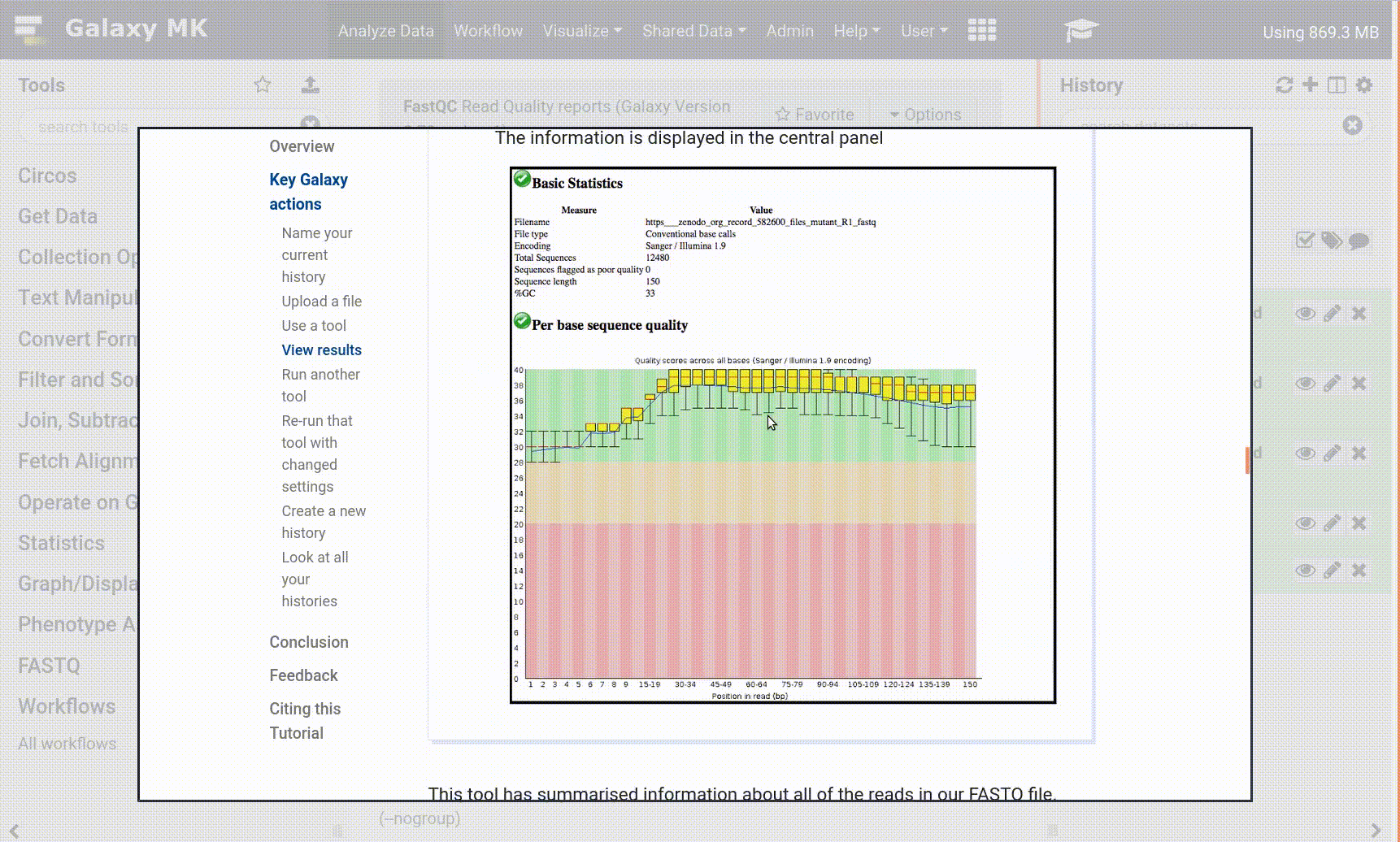
Praktische Übung: Dateiinhalt anzeigen
Klicken Sie auf das galaxy-eye (Auge)-Symbol (Daten anzeigen) der Peak-Datei, um den Inhalt der Datei anzuzeigen
Es sollte wie folgt aussehen:
Ansicht des Inhalts der Regionen der Gene von UCSC
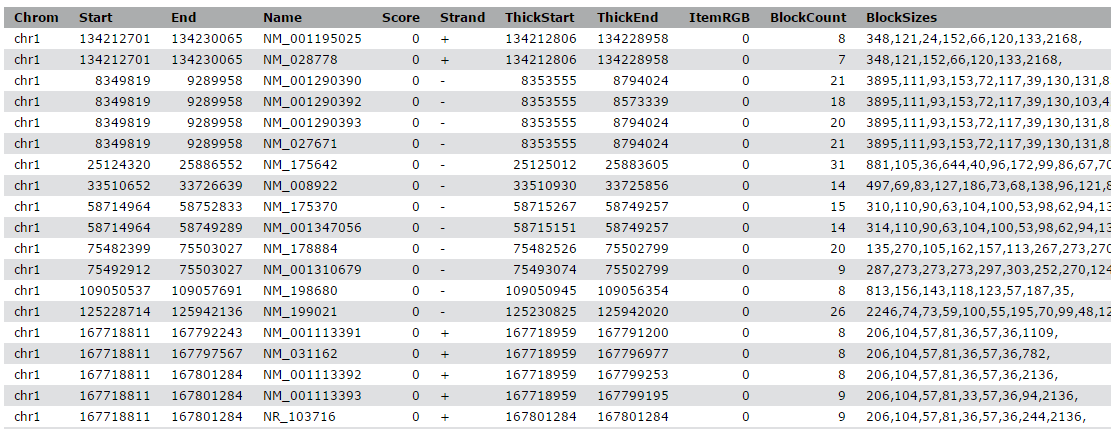
FrageWährend die Datei von UCSC Bezeichnungen für die Spalten hat, hat die Peak-Datei keine. Können Sie erraten, wofür die Spalten stehen?
Diese Peak-Datei hat kein Standardformat, und allein durch ihre Betrachtung können wir nicht herausfinden, was die Zahlen in den verschiedenen Spalten bedeuten. In der Veröffentlichung erwähnen die Autoren, dass sie den Peak Caller HPeak verwendet haben.
Aus dem HPeak-Handbuch geht hervor, dass die Spalten die folgenden Informationen enthalten:
- Chromosomenname nach Nummer
- Startkoordinate
- Endkoordinate
- Länge
- Stelle innerhalb des Peaks, die die höchste hypothetische DNA-Fragmentabdeckung aufweist (Gipfel)
- nicht relevant
- nicht relevant
Um die beiden Dateien zu vergleichen, müssen wir sicherstellen, dass die
Chromosomennamen das gleiche Format haben. Wie wir sehen können, fehlt in der Peak-Datei
das chr vor jeder Chromosomennummer. Aber was passiert mit Chromosom 20 und 21? Werden
sie stattdessen X und Y heißen? Schauen wir nach:
Praktische Übung: Ende der Datei anzeigen
- Suche nach Select last lines from a dataset (tail) ( Galaxy version 9.3+galaxy1) tool und führe es mit den folgenden Einstellungen aus:
- “Textdatei “: unsere Peakdatei
GSE37268_mof3.out.hpeak.txt.gz- “Operation “:
Keep last lines- “Anzahl der Zeilen “: Wählen Sie einen Wert, z.B.
100- Klick auf Werkzeug starten
- Warten, bis der Auftrag beendet ist
Untersuchen Sie die Datei über das galaxy-eye (Auge) Symbol (Daten ansehen)
Frage
- Wie sind die Chromosomen benannt?
- Wie sind die Chromosomen X und Y benannt?
- Die Chromosomen werden einfach durch ihre Nummer angegeben. In der Gendatei von UCSC beginnen sie mit
chr- Die Chromosomen X und Y werden 20 und 21 genannt
Um die Chromosomennamen zu konvertieren, müssen wir also zwei Dinge tun:
chrhinzufügen- 20 und 21 in X und Y ändern
Praktische Übung: Anpassen der Chromosomennamen
- Replace Text ( Galaxy version 1.1.3) in einer bestimmten Spalte mit den folgenden Einstellungen:
- “Zu verarbeitende Datei “: unsere Peak-Datei
GSE37268_mof3.out.hpeak.txt.gz- “in Spalte “:
1“Muster suchen “:
[0-9]+Es wird nach numerischen Ziffern gesucht
“Ersetzen durch “:
chr&
&ist ein Platzhalter für das Suchergebnis der MustersucheBenennen Sie Ihre Ausgabedatei in
chr prefix addedum.- Replace Text ( Galaxy version 1.1.3) : Führen wir das Tool mit zwei weiteren Ersetzungen erneut aus
- “Zu verarbeitende Datei “: die Ausgabe des letzten Laufs,
chr prefix added- “in Spalte “:
1- param-repeat Ersetzen
- “Muster suchen “:
chr20- “Ersetzen durch “:
chrX- param-repeat Ersetzen einfügen
- “Muster suchen “:
chr21- “Ersetzen durch “:
chrY
- Erweitern Sie die Datensatzinformationen
- Drücken Sie das Symbol galaxy-refresh (Diesen Job erneut ausführen)
Prüfen Sie die letzte Datei über das galaxy-eye (Auge) Symbol. Waren wir erfolgreich?
Wir haben jetzt eine ganze Reihe von Dateien und müssen bei jedem Schritt darauf achten, die richtigen auszuwählen.
FrageWie viele Regionen befinden sich in unserer Ausgabedatei? Sie können auf den Namen der Ausgabedatei klicken, um sie zu erweitern und die Anzahl zu sehen.
Sie sollte der Anzahl der Regionen in Ihrer ersten Datei,
GSE37268_mof3.out.hpeak.txt.gz, entsprechen: 48.647 Wenn in Ihrer Datei 100 Regionen angezeigt werden, haben Sie das Programm mit der DateiTailausgeführt und müssen die Schritte erneut ausführen.- Umbenennen der Datei in etwas besser Erkennbares, z.B.
Peak regions
Analyse
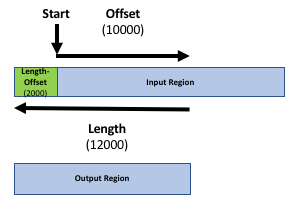
Unser Ziel ist es, die beiden Regionsdateien (die Gendatei und die Peakdatei aus der Veröffentlichung) zu vergleichen, um herauszufinden, welche Peaks mit welchen Genen verbunden sind. Wenn Sie nur wissen wollen, welche Peaks sich innerhalb von Genen (innerhalb des Genkörpers) befinden, können Sie den nächsten Schritt überspringen. Andernfalls könnte es sinnvoll sein, die Promoter-Region der Gene in den Vergleich einzubeziehen, z. B. weil Sie Transkriptionsfaktoren in ChIP-seq-Experimente einbeziehen wollen. Es gibt keine strenge Definition für die Promotorregion, aber 2kb stromaufwärts vom TSS (Beginn der Region) wird üblicherweise verwendet. Wir verwenden das Tool Get Flanks, um Regionen zu erhalten, die 2kb Basen stromaufwärts vom Start des Gens bis 10kb Basen stromabwärts vom Start liegen (12kb Länge). Dazu teilen wir dem Tool Get Flanks mit, dass wir Regionen vor dem Start mit einem Offset von 10kb suchen, die 12kb lang sind, wie im folgenden Diagramm dargestellt.
Praktische Übung: Promoterregion zu Gendatensätzen hinzufügen
- Get Flanks ( Galaxy version 1.0.0) gibt flankierende Region/en für jedes Gen zurück, mit den folgenden Einstellungen:
- “Daten auswählen “:
GenesDatei von UCSC- “Region “:
Around Start- “Lage der flankierenden Region/en “:
Upstream- “Versatz “:
10000- “Länge der flankierenden Region(en) “:
12000Dieses Tool liefert flankierende Regionen für jedes Gen
Vergleichen Sie die Zeilen der resultierenden BED-Datei mit der Eingabe, um herauszufinden, wie sich die Start- und Endpositionen geändert haben
Klicken Sie auf Scratchbook aktivieren/deaktivieren in der oberen Leiste
- Klicken Sie auf das galaxy-eye (Auge)-Symbol der zu prüfenden Dateien
Klicken Sie auf Scratchbook anzeigen/ausblenden
- Benennen Sie Ihren Datensatz so um, dass er Ihre Ergebnisse widerspiegelt (
Promoter regions)

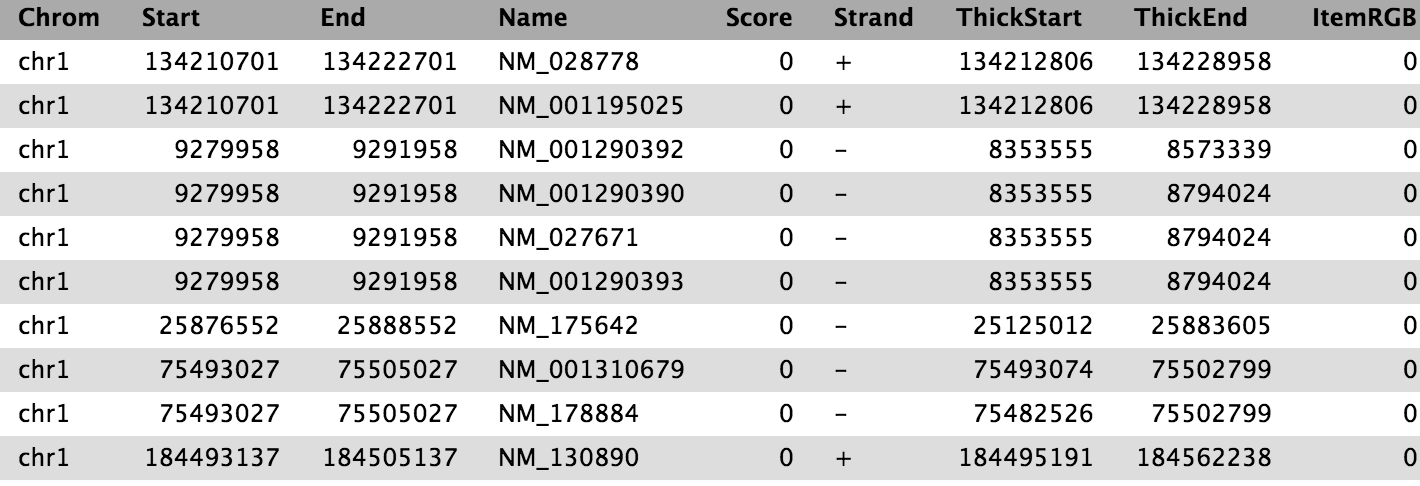
Die Ausgabe besteht aus Regionen, die 2kb stromaufwärts vom TSS beginnen und 10kb stromabwärts umfassen. Für Eingaberegionen auf dem positiven Strang, z. B. chr1 134212701 134230065, ergibt dies chr1 134210701 134222701. Für Regionen auf dem negativen Strang, z. B. chr1 8349819 9289958, ergibt dies chr1 9279958 9291958.
Sie haben vielleicht bemerkt, dass die UCSC-Datei im Format BED vorliegt und mit einer
Datenbank verknüpft ist. Genau das wollen wir auch für unsere Peak-Datei. Das Tool
Intersect, das wir verwenden werden, kann Intervalldateien automatisch in das
BED-Format konvertieren, aber wir werden unsere Intervalldatei hier explizit
konvertieren, um zu zeigen, wie dies mit Galaxy erreicht werden kann.
Praktische Übung: Format und Datenbank ändern
- Klicken Sie auf das galaxy-pencil (Bleistift)-Symbol im History-Eintrag unserer Peak-Region-Datei
- Wechsel zur Registerkarte Datentypen
- Im Abschnitt In Datentyp konvertieren unter “Zieldatentyp “ wählen:
bed (Verwendung 'Convert Genomic Interval To Bed')- Drücken Sie Datensatz erstellen
- Prüfen Sie, ob die “Datenbank/Build”
mm9ist (die in der Publikation verwendete Datenbank-Build für Mäuse)- Benennen Sie die Datei erneut in etwas Erkennbares um, z.B.
Peak regions BED
Es ist an der Zeit, die überlappenden Intervalle zu finden (endlich!). Dazu wollen wir die Gene extrahieren, die sich mit unseren Peaks überlappen/überschneiden.
Praktische Übung: Überschneidungen finden
- Intersect ( Galaxy version 1.0.0) die Intervalle zweier Datensätze, mit den folgenden Einstellungen:
- “Rückgabe “:
Overlapping Intervals- “von “: die UCSC-Datei mit Promotorregionen (
Promoter regions)- “die sich schneiden “: unsere Peak-Region-Datei von Ersetzen (
Peak regions BED)- “für mindestens “:
1KommentarDie Reihenfolge der Eingaben ist wichtig! Wir wollen am Ende eine Liste von Genen erhalten, also muss der entsprechende Datensatz mit den Geninformationen die erste Eingabe sein (
Promoter regions).
Wir haben jetzt die Liste der Gene (Spalte 4), die sich mit den Peak-Regionen überschneiden, ähnlich wie oben gezeigt.
Um einen besseren Überblick über die erhaltenen Gene zu bekommen, wollen wir uns ihre Verteilung auf die verschiedenen Chromosomen ansehen. Wir gruppieren die Tabelle nach Chromosomen und zählen die Anzahl der Gene mit Peaks auf jedem Chromosom
Praktische Übung: Gene auf verschiedenen Chromosomen zählen
- Gruppieren Sie Daten nach einer Spalte und führen Sie eine Aggregationsoperation für andere Spalten durch, mit den folgenden Einstellungen.
- “Daten auswählen “ zum Ergebnis der Schnittmenge
- “Gruppieren nach Spalte “:
Column 1- Drücken Sie Einfügen Operation und wählen Sie:
- “Typ”:
Count- “In der Spalte “:
Column 1- “Ergebnis auf die nächste ganze Zahl runden “:
NoFrageWelches Chromosom enthielt die höchste Anzahl von Zielgenen?
Das Ergebnis variiert bei verschiedenen Einstellungen, z. B. kann sich die Annotation aufgrund von Aktualisierungen bei UCSC ändern. Wenn Sie Schritt für Schritt mit der gleichen Annotation vorgehen, sollte das Ergebnis Chromosom 11 mit 2164 Genen sein. Beachten Sie, dass Sie aus Gründen der Reproduzierbarkeit alle in der Analyse verwendeten Eingabedaten aufbewahren sollten. Ein erneuter Durchlauf der Analyse mit demselben Satz von Parametern, die in Galaxy gespeichert sind, kann zu einem anderen Ergebnis führen, wenn sich die Eingaben geändert haben, z. B. die Annotation von UCSC.
Visualisierung
Wir haben einige schöne aggregierte Daten, warum also nicht einen Barchart davon zeichnen?
Bevor wir das tun, sollten wir unsere gruppierten Daten noch ein wenig aufpolieren.
Sie haben vielleicht bemerkt, dass die Mäusechromosomen in diesem Datensatz nicht in der richtigen Reihenfolge aufgelistet sind (das Tool Group versuchte, sie zu sortieren, tat dies aber in alphabetischer Reihenfolge).
Wir können dies beheben, indem wir ein spezielles Tool zum Sortieren unserer Daten verwenden.
Praktische Übung: Sortierreihenfolge der Genanzahltabelle korrigieren
- Sort ( Galaxy version 1.1.1) Daten in aufsteigender oder absteigender Reihenfolge, mit den folgenden Einstellungen:
- “Sortierabfrage “: Ergebnis der Ausführung des Gruppentools
- in param-repeat “Spaltenselektionen “
- “in Spalte “:
Column 1- “in “:
Ascending order- “Flavor “:
Natural/Version sort (-V)Manchmal gibt es mehrere Werkzeuge mit sehr ähnlichen Namen. Wenn die Parameter im Tutorial nicht mit dem übereinstimmen, was Sie in Galaxy sehen, versuchen Sie bitte das Folgende:
Verwenden Sie den Tutorial Mode curriculum in Galaxy, und klicken Sie auf den blauen Tool-Button im Tutorial, um automatisch das richtige Tool und die richtige Version zu öffnen (noch nicht für alle Tutorials verfügbar)
Die Tools werden häufig auf neue Versionen aktualisiert. Auf Ihrem Galaxy sind möglicherweise mehrere Versionen desselben Tools verfügbar. Standardmäßig wird Ihnen die neueste Version des Tools angezeigt. Dies ist möglicherweise NICHT dasselbe Tool, das in dem von Ihnen aufgerufenen Lernprogramm verwendet wird. Wenn Sie in einem Schritt ein neueres Tool verwenden und im nächsten Schritt ein älteres Tool benutzen, kann dies fehlschlagen! Um sicher zu gehen, dass Sie die gleiche Werkzeugversion eines bestimmten Tutorials verwenden, benutzen Sie die Funktion Tutorial mode.
- Öffnen Sie Ihren Galaxy-Server
- Klicken Sie auf das Symbol curriculum im oberen Menü, um das GTN in Galaxy zu öffnen.
- Navigieren Sie zu Ihrem Lernprogramm
- Werkzeugnamen in Tutorials sind blaue Schaltflächen, die das richtige Werkzeug für Sie öffnen
- Hinweis: dies funktioniert (noch) nicht bei allen Tutorials
- Sie können auf eine beliebige Stelle in dem ausgegrauten Bereich außerhalb des Lernprogramms klicken, um zur analytischen Galaxy-Oberfläche zurückzukehren
Warnung: Nicht alle Browser funktionieren!
- Es gab einige Probleme mit dem Lernprogramm-Modus in Safari für Mac-Benutzer.
- Versuchen Sie einen anderen Browser, wenn Sie die Schaltfläche nicht sehen.
Überprüfen Sie, ob der gesamte Werkzeugname mit dem im Tutorial übereinstimmt. Bitte überprüfen Sie das:
Vollständiger Werkzeugname:
Daten in aufsteigender oder absteigender Reihenfolge sortierenWerkzeugversion:
1.1.1(wird hinter den Werkzeugnamen geschrieben)
Großartig, wir sind bereit, Dinge zu zeichnen!
Praktische Übung: Balkendiagramm zeichnen
- Klicken Sie auf galaxy-barchart (visualisieren) auf die Ausgabe des Werkzeugs Sortieren
- Wähle
Bar diagram (NVD3)- Klicken Sie auf das Symbol « in der oberen rechten Ecke
- Wählen Sie einen Titel bei Angeben eines Titels, z.B.
Gene counts per chromosome- Wechseln Sie zum galaxy-chart-select-data Daten auswählen
- Spielen Sie mit den Einstellungen herum
Wenn Sie zufrieden sind, klicken Sie auf das galaxy-save Speichern Visualisierung oben rechts im Hauptfenster
Damit wird die Datei in Ihren gespeicherten Visualisierungen gespeichert. Später können Sie sie unter Daten -> Visualisierungen im oberen Menü von Galaxy ansehen, herunterladen oder mit anderen teilen.
Arbeitsablauf extrahieren
Wenn Sie sich Ihren Verlauf genau ansehen, können Sie feststellen, dass er alle Schritte unserer Analyse enthält, vom Anfang bis zum Ende. Durch die Erstellung dieses Verlaufs haben wir tatsächlich eine vollständige Aufzeichnung unserer Analyse mit Galaxy erstellt, in der alle Parametereinstellungen, die bei jedem Schritt angewendet wurden, erhalten bleiben. Wäre es nicht schön, diesen Verlauf einfach in einen Workflow umzuwandeln, den wir immer wieder ausführen können?
Galaxy macht dies mit der Option Extract workflow sehr einfach. Das bedeutet, dass Sie
jedes Mal, wenn Sie einen Arbeitsablauf erstellen wollen, diesen einmal manuell
durchführen und dann in einen Arbeitsablauf umwandeln können, so dass es beim nächsten
Mal viel weniger Arbeit ist, die gleiche Analyse durchzuführen. Außerdem können Sie Ihre
Analyse problemlos weitergeben oder veröffentlichen.
Praktische Übung: Workflow extrahieren
Aufräumen: Entfernen Sie alle fehlgeschlagenen (roten) Aufträge aus Ihrer Historie, indem Sie auf die Schaltfläche galaxy-delete klicken.
Dies wird die Erstellung des Arbeitsablaufs erleichtern.
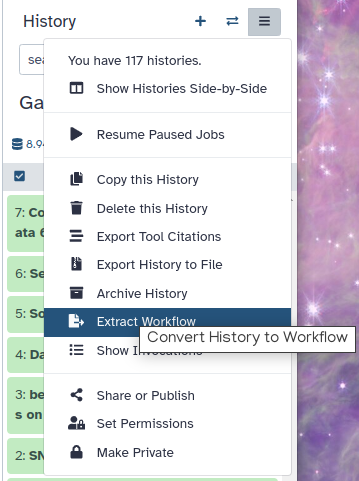
Klicken Sie auf galaxy-gear (Verlaufsoptionen) am oberen Rand des Verlaufsfensters und wählen Sie Workflow extrahieren.
Das zentrale Bedienfeld zeigt den Inhalt der Historie in umgekehrter Reihenfolge an (die älteste Datei steht oben), und Sie können auswählen, welche Schritte in den Arbeitsablauf aufgenommen werden sollen.
Ersetzen Sie den Arbeitsablaufnamen durch etwas Beschreibenderes, zum Beispiel:
From peaks to genesWenn es Schritte gibt, die nicht in den Arbeitsablauf einbezogen werden sollen, können Sie sie in der ersten Spalte der Kästchen entmarkieren.
Da wir einige Schritte durchgeführt haben, die spezifisch für unsere benutzerdefinierte Peak-Datei waren, möchten wir sie vielleicht ausschließen:
- Letztes auswählen tool
- alle Ersetzen von Text tool steps
- Genomische Intervalle in BED umwandeln
- Flanken holen tool
Klicken Sie auf die Schaltfläche Workflow erstellen am oberen Rand.
Sie erhalten eine Meldung, dass der Workflow erstellt wurde. Aber wo ist er geblieben?
Klicken Sie auf Workflow im linken Menü von Galaxy
Hier haben Sie eine Liste aller Ihrer Arbeitsabläufe
Wählen Sie den neu erstellten Workflow aus und klicken Sie auf Bearbeiten
Sie sollten etwas ähnliches wie das hier sehen:
Kommentar: Der Workflow-EditorWir können den Workflow im Workflow-Editor von Galaxy untersuchen. Hier können Sie die Parametereinstellungen der einzelnen Schritte anzeigen/ändern, Werkzeuge hinzufügen und entfernen und die Ausgabe eines Werkzeugs mit der Eingabe eines anderen verbinden - alles auf einfache und grafische Weise. Sie können diesen Editor auch verwenden, um Workflows von Grund auf neu zu erstellen.
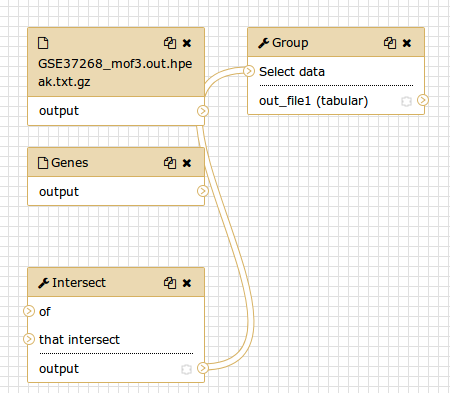
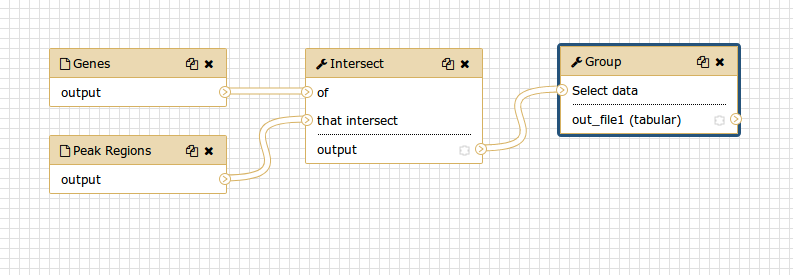
Obwohl wir unsere beiden Eingaben im Workflow haben, fehlt ihnen die Verbindung zum ersten Werkzeug (Intersect tool), da wir einige der Zwischenschritte nicht übernommen haben.
- Verbinden Sie jeden Eingabedatensatz mit dem Tool Intersect tool, indem Sie den nach außen zeigenden Pfeil auf der rechten Seite des Kästchens (das eine Ausgabe bezeichnet) auf einen nach innen zeigenden Pfeil auf der linken Seite des Intersect-Kästchens (das eine Eingabe bezeichnet) ziehen
- Umbenennung der Eingabedatensätze in
Reference regionsundPeak regions- Drücken Sie Auto Re-layout, um unsere Ansicht aufzuräumen
- Klicken Sie auf das galaxy-save Speichern (oben), um Ihre Änderungen zu speichern
Bei der Ausführung eines Workflows ist der Benutzer in der Regel in erster Linie an dem Endprodukt und nicht an allen Zwischenschritten interessiert. Standardmäßig werden alle Ausgaben eines Workflows angezeigt, aber wir können Galaxy explizit mitteilen, welche Ausgaben für einen bestimmten Workflow angezeigt und welche ausgeblendet werden sollen. Dieses Verhalten wird durch das kleine Sternchen neben jedem Ausgabedatensatz gesteuert:
Wenn Sie auf dieses Sternchen für einen der Ausgabedatensätze klicken, werden nur die Dateien mit einem Sternchen angezeigt, und alle Ausgaben ohne Sternchen werden ausgeblendet (Beachten Sie, dass das Anklicken von allen Ausgaben denselben Effekt hat wie das Anklicken von keiner der Ausgaben, in beiden Fällen werden alle Datensätze angezeigt).
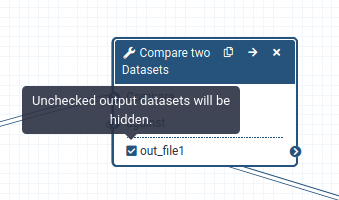
Nun ist es an der Zeit, unseren Arbeitsablauf für einen anspruchsvolleren Ansatz wiederzuverwenden.
Teil 2: Anspruchsvollerer Ansatz
In Teil 1 haben wir eine Überlappungsdefinition von 1 bp (Standardeinstellung) verwendet, um Gene zu identifizieren, die mit den Peakregionen verbunden sind. Um eine aussagekräftigere Definition zu erhalten, könnten wir stattdessen die Gene identifizieren, die sich dort überlappen, wo die meisten Reads konzentriert sind, den Peak-Gipfel. Wir verwenden die Informationen über die Position des Peak-Gipfels, die in der ursprünglichen Peak-Datei enthalten sind, und prüfen, ob sich die Gipfel mit Genen überschneiden.
Vorbereitung
Wir brauchen wieder unsere Peak-Datei, aber wir möchten in einem sauberen Verlauf arbeiten. Anstatt sie zweimal hochzuladen, können wir sie in einen neuen Verlauf kopieren.
Praktische Übung: Historische Elemente kopieren
Erstellen Sie einen neuen Verlauf und geben Sie ihm einen neuen Namen wie
Galaxy Introduction Part 2Um einen neuen Verlauf zu erstellen, klicken Sie einfach auf das Symbol new-history am oberen Rand des Verlaufsfensters:
Klicken Sie auf die Historienoptionen oben rechts in Ihrer Historie. Klicken Sie auf die Option Verläufe nebeneinander anzeigen
Sie sollten nun beide Historien nebeneinander sehen
- Ziehen Sie die bearbeitete Peak-Datei (
Peak regions, nach den Ersetzungsschritten), die die Gipfelinformationen enthält, in Ihren neuen Verlauf.- Klicken Sie auf
Galaxyin der oberen Menüleiste (oben links), um zu Ihrem Analysefenster zurückzukehren
Peak-Gipfel-Datei erstellen
Wir müssen eine neue BED-Datei aus der ursprünglichen Peak-Datei erzeugen, die die
Positionen der Peak-Gipfel enthält. Der Beginn des Gipfels ist der Beginn des Peaks
(Spalte 2) plus die Stelle innerhalb des Peaks, die die höchste hypothetische
DNA-Fragmentabdeckung aufweist (Spalte 5, abgerundet auf die nächstkleinere ganze Zahl,
da einige Peakgipfel zwischen zwei Basen liegen). Als Ende der Peakregion definieren wir
einfach start + 1.
Praktische Übung: Gipfeldatei erstellen
- Compute on rows ( Galaxy version 2.0) mit den folgenden Parametern:
- “Eingabedatei “: unsere Peakdatei
Peak regions(die Intervallformatdatei)- *“Eingabe hat eine Kopfzeile mit Spaltennamen?”:
No- In “Ausdrücke “:
- param-repeat “Ausdrücke “
- “Ausdruck hinzufügen “:
c2 + int(c5)- “Art der Operation “: Anhängen
- param-repeat “Ausdrücke “
- “Ausdruck hinzufügen “:
c8 + 1- “Art der Operation “: Anhängen
Dies wird eine 8. und eine 9. Spalte in unserer Tabelle erzeugen, die wir im nächsten Schritt verwenden werden:
- Umbenennen der Ausgabe
Peak summit regions
Jetzt schneiden wir nur das Chromosom sowie den Anfang und das Ende des Gipfels aus:
Praktische Übung: Spalten ausschneiden
- Cut Spalten aus einer Tabelle mit den folgenden Einstellungen:
- “Spalten ausschneiden “:
c1,c8,c9- “Getrennt durch Tabulator “:
Tab- “Von “:
Peak summit regionsDie Ausgabe von Cut wird im Format
tabularerfolgen.
Ändern Sie das Format in
interval(verwenden Sie das galaxy-pencil), da dies das ist, was das Tool Intersect erwartet.
- Klicken Sie auf das galaxy-pencil Bleistift-Symbol für den Datensatz, um seine Attribute zu bearbeiten
- Klicken Sie im zentralen Panel auf galaxy-chart-select-data *registerkarte *Datentypen** oben
- Im Feld galaxy-chart-select-data Datentyp zuweisen, wählen Sie
intervalaus dem “Neuer Typ“-Dropdown
- Tipp: Sie können mit der Eingabe des Datentyps in das Feld beginnen, um das Dropdown-Menü zu filtern
- Klicken Sie auf die Schaltfläche Speichern
Die Ausgabe sollte wie folgt aussehen:
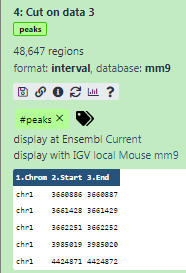
Gennamen holen
Die RefSeq-Gene, die wir von der UCSC heruntergeladen haben, enthielten nur die RefSeq-Kennungen, aber nicht die Gennamen. Um am Ende eine Liste der Gennamen zu erhalten, verwenden wir eine andere BED-Datei aus den Datenbibliotheken.
KommentarEs gibt mehrere Möglichkeiten, die Gennamen einzugeben, wenn Sie dies selbst tun müssen. Eine Möglichkeit besteht darin, ein Mapping über Biomart abzurufen und dann die beiden Dateien zu verbinden (Zwei Datensätze nebeneinander auf einem bestimmten Feld verbinden tool). Eine andere Möglichkeit ist, die vollständige RefSeq-Tabelle von UCSC zu erhalten und sie manuell in das BED-Format zu konvertieren.
Praktische Übung: Daten-Upload
Import
mm9.RefSeq_genes_from_UCSC.bedvon Zenodo oder aus der Datenbibliothek:https://zenodo.org/record/1025586/files/mm9.RefSeq_genes_from_UCSC.bed
- Kopieren der Linkposition
Klicken Sie auf galaxy-upload Daten hochladen am oberen Rand der Werkzeugleiste
- Wählen Sie galaxy-wf-edit Daten einfügen/holen
Fügen Sie den/die Link(s) in das Textfeld ein
Genom auf
mm9ändernDrücken Sie Start
- Schließen Sie das Fenster
Als Alternative zum Hochladen der Daten von einer URL oder Ihrem Computer können die Dateien auch von einer Shared Data Library zur Verfügung gestellt werden:
- Gehen Sie in Bibliotheken (linker Bereich)
Navigieren Sie zu : Klicken Sie auf “GTN - Material”, “Introduction to Galaxy Analyses”, “From peaks to genes”, und dann “DOI: 10.5281/zenodo.1025586” oder dem richtigen Ordner, wie von Ihrem Ausbilder angegeben.
- Wählen Sie die gewünschten Dateien aus
- Klicken Sie auf Zur Historie hinzufügen galaxy-dropdown am oberen Rand und wählen Sie as Datasets aus dem Dropdown-Menü
Wählen Sie im Pop-up-Fenster
- “Historie auswählen “: die Historie, in die Sie die Daten importieren möchten (oder erstellen Sie eine neue)
- Klicken Sie auf Importieren
Standardmäßig nimmt Galaxy den Link als Namen, also benennen Sie sie um.
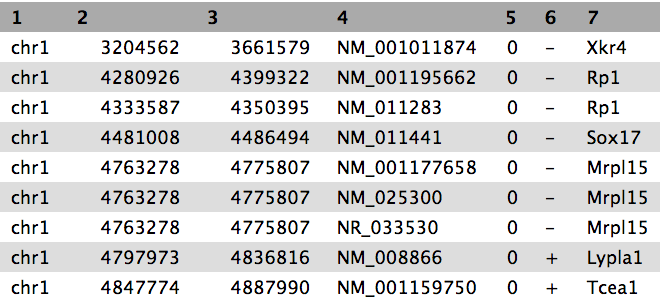
Überprüfen Sie den Inhalt der Datei, um zu sehen, ob sie Gennamen enthält. Er sollte ähnlich wie unten aussehen:
- Umbenennen in
mm9.RefSeq_genes- Anwenden des Tags
#genes
Arbeitsablauf wiederholen
Es ist an der Zeit, den zuvor erstellten Arbeitsablauf wiederzuverwenden.
Praktische Übung: Ausführen eines Arbeitsablaufs
- Öffnen Sie das Workflow-Menü (linke Menüleiste)
- Suchen Sie den Workflow, den Sie im vorherigen Abschnitt erstellt haben, und wählen Sie die Option Ausführen
- Wählen Sie als Eingaben unsere
mm9.RefSeq_genes(#genes) BED-Datei und das Ergebnis des Cut-Tools (#peaks)Klick Workflow starten
Die Ausgaben sollten in der Historie erscheinen, aber es kann einige Zeit dauern, bis sie fertig sind.
Wir haben unseren Workflow verwendet, um unsere Analyse mit den Spitzenwerten zu wiederholen. Das Tool Group lieferte wieder eine Liste mit der Anzahl der in jedem Chromosom gefundenen Gene. Aber wäre es nicht interessanter, die Anzahl der Peaks in jedem einzelnen Gen zu kennen? Lassen Sie uns den Arbeitsablauf mit anderen Einstellungen wiederholen!
Praktische Übung: Ausführen eines Arbeitsablaufs mit geänderten Einstellungen
- Öffnet das Workflow-Menü (linke Menüleiste)
- Suchen Sie den Workflow, den Sie im vorherigen Abschnitt erstellt haben
- Wählen Sie die Option Ausführen
- Wählen Sie als Eingaben unsere
mm9.RefSeq_genes(#genes) BED-Datei und das Ergebnis des Cut-Tools (#peaks)- Klicken Sie auf den Titel des Werkzeugs tool Gruppenwerkzeugs, um die Optionen zu erweitern.
- Ändern Sie die folgenden Einstellungen durch Klicken auf das galaxy-pencil (Bleistift)-Symbol auf der linken Seite:
- “Gruppieren nach Spalte “:
7- In “Operation “:
- “In der Spalte “:
7- Klick Workflow starten
Herzlichen Glückwunsch! Sie sollten nun eine Datei mit allen eindeutigen Gennamen und der Anzahl der darin enthaltenen Peaks haben.
FrageDie Liste der eindeutigen Gene ist nicht sortiert. Versuchen Sie, sie selbst zu sortieren!
Sie können das Werkzeug “Daten in aufsteigender oder absteigender Reihenfolge Sortieren ( Galaxy version 9.5+galaxy2)” auf Spalte 2 und “schnelle numerische Sortierung” verwenden.
Teilen Sie Ihre Arbeit
Eine der wichtigsten Funktionen von Galaxy kommt am Ende einer Analyse. Wenn Sie beeindruckende Ergebnisse veröffentlicht haben, ist es wichtig, dass andere Forscher in der Lage sind, Ihr In-Silico-Experiment zu reproduzieren. Galaxy ermöglicht es den Nutzern, ihre Arbeitsabläufe und Historien mit anderen zu teilen.
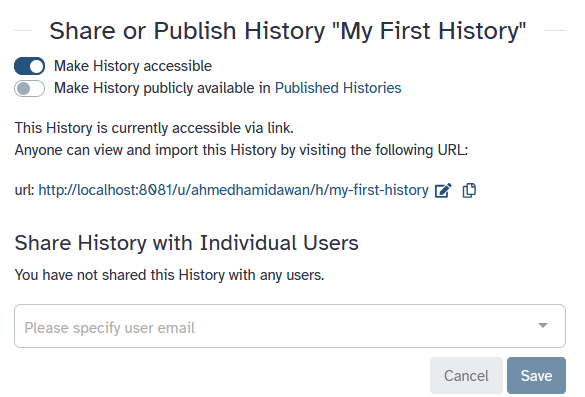
Um einen Verlauf zu teilen, klicken Sie auf die galaxy-history-options
Verlaufsoptionen und wählen Sie Share or Publish. Auf dieser Seite können Sie 3 Dinge
tun:
-
Zugänglich machen über Link
Dies erzeugt einen Link, den Sie an andere weitergeben können. Jeder, der diesen Link hat, kann Ihren Verlauf sehen.
-
Historie öffentlich zugänglich machen in Published Histories
Damit wird nicht nur ein Link erstellt, sondern auch Ihr Verlauf veröffentlicht. Das bedeutet, dass Ihr Verlauf unter
Data → Histories → Published Historiesim oberen Menü aufgeführt wird. -
Mit einzelnen Benutzern teilen
Dadurch wird der Verlauf nur für bestimmte Benutzer der Galaxy-Instanz freigegeben.
Praktische Übung: History und Arbeitsablauf teilen
- Teilen Sie eine Ihrer Historien mit Ihrem Nachbarn
- Schau mal, ob du das auch mit deinem Arbeitsablauf machen kannst!
Finde die History und/oder den Arbeitsablauf, den dein Nachbar teilt
Historien, die für bestimmte Benutzer freigegeben sind, können von diesen Benutzern unter
Data → Histories → Histories shared with meeingesehen werden.
Schlussfolgerung
trophy Sie haben gerade Ihre erste Analyse in Galaxy durchgeführt. Sie haben auch einen Arbeitsablauf für Ihre Analyse erstellt, so dass Sie dieselbe Analyse leicht mit anderen Datensätzen wiederholen können. Außerdem haben Sie Ihre Ergebnisse und Methoden mit anderen geteilt.
Du hast das Tutorial abgeschlossen
Zusammenfassung
Galaxy bietet eine benutzerfreundliche grafische Oberfläche für oft komplexe Kommandozeilen‑Werkzeuge
Galaxy hält eine vollständige Aufzeichnung Ihrer Analyse in einer History
Workflows ermöglichen es, Ihre Analyse auf verschiedene Daten zu wiederholen
Galaxy kann sich mit externen Quellen zur Datenimport‑ und Visualisierungszwecken verbinden
Galaxy bietet Möglichkeiten, Ihre Ergebnisse und Methoden mit anderen zu teilen
Häufig gestellte Fragen
Fragen zu diesem Tutorial? Schau auf die verfügbaren FAQ-Seiten und Support-KanäleFörderung
- Li, X., L. Li, R. Pandey, J. S. Byun, K. Gardner et al., 2012 The Histone Acetyltransferase MOF Is a Key Regulator of the Embryonic Stem Cell Core Transcriptional Network. Cell Stem Cell 11: 163–178. 10.1016/j.stem.2012.04.023
Feedback
Feedback Möglichkeit für Lehrende: Wie lief dein Kurs.
Für Kursteilnehmer/-innen oder Studierende: Über Rückmeldung durch das untenstehende Formular würden wir uns freuen.

Dieses Tutorial zitieren
- Anne Pajon, Clemens Blank, Bérénice Batut, Björn Grüning, Nicola Soranzo, Dilmurat Yusuf, Sarah Peter, Helena Rasche, Von Peaks zu Genen (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/introduction/tutorials/galaxy-intro-peaks2genes/tutorial_DE.html Online; accessed TODAY
- Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{introduction-galaxy-intro-peaks2genes, author = "Anne Pajon and Clemens Blank and Bérénice Batut and Björn Grüning and Nicola Soranzo and Dilmurat Yusuf and Sarah Peter and Helena Rasche", title = "Von Peaks zu Genen (Galaxy Training Materials)", year = "", month = "", day = "", url = "\url{https://training.galaxyproject.org/training-material/topics/introduction/tutorials/galaxy-intro-peaks2genes/tutorial_DE.html}", note = "[Online; accessed TODAY]" } @article{Hiltemann_2023, doi = {10.1371/journal.pcbi.1010752}, url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752}, year = 2023, month = {jan}, publisher = {Public Library of Science ({PLoS})}, volume = {19}, number = {1}, pages = {e1010752}, author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and}, editor = {Francis Ouellette}, title = {Galaxy Training: A powerful framework for teaching!}, journal = {PLoS Comput Biol} }
Förderung
Diese Personen oder Organisationen unterstützten die Entwicklung dieser Quelle finanziell.
Herzlichen Glückwunsch zum erfolgreichen Abschluss dieses Tutorials!

You can use Ephemeris's
shed-tools installcommand to install the tools used in this tutorial.shed-tools install [-g GALAXY] [-a API_KEY] -t <(curl https://training.galaxyproject.org/training-material/api/topics/introduction/tutorials/galaxy-intro-peaks2genes/tutorial.json | jq .admin_install_yaml -r)Alternatively you can copy and paste the following YAML
--- install_tool_dependencies: true install_repository_dependencies: true install_resolver_dependencies: true tools: - name: text_processing owner: bgruening revisions: d698c222f354 tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: text_processing owner: bgruening revisions: c41d78ae5fee tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: text_processing owner: bgruening revisions: c41d78ae5fee tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: text_processing owner: bgruening revisions: d698c222f354 tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: text_processing owner: bgruening revisions: c41d78ae5fee tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: column_maker owner: devteam revisions: aff5135563c6 tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: get_flanks owner: devteam revisions: 077f404ae1bb tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: intersect owner: devteam revisions: 69c10b56f46d tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/