Understanding Barcodes
| Author(s) |
|
| Editor(s) |
|
| Reviewers |
|
OverviewQuestions:
Objectives:
What are barcodes?
What is their purpose?
How are barcodes formatted?
Requirements:
Demultiplex FASTQ data via UMI-tools
Understand and validate the extraction of barcodes
Time estimation: 2 hoursSupporting Materials:Published: Feb 20, 2019Last modification: Jun 25, 2026License: Tutorial Content is licensed under Creative Commons Attribution 4.0 International License. The GTN Framework is licensed under MITpurl PURL: https://gxy.io/GTN:T00256rating Rating: 4.2 (0 recent ratings, 27 all time)version Revision: 12
Barcodes are small oligonucleotides that are inserted into the captured sequence at a specific point, and provide two pieces of information about the sequence:
- Which cell the sequence came from.
- Which transcript the sequence came from
When the sequence is mapped against a reference genome, we can then see which gene locus it aligns to and qualitatively assert that, together with the two pieces of information above, the sequence depicts a transcript from a specific gene that originated from a specific cell.
Barcodes come in a variety of formats, and in this tutorial we will be looking at the CEL-Seq2 protocol Hashimshony et al. 2016 used in droplet-based single-cell RNA-seq.
The CEL-Seq2 Protocol
CEL-Seq2 is a paired-end protocol, meaning that two primers bind to opposite ends of a cDNA strand in order to sequence. Each primer has a specific role.
In this case; Read1 contains the barcoding information followed by the polyT tail of the messenger RNA, and Read2 contains the actual sequence. Here, Read1 is regarded as the ‘forward’ strand and Read2 as the ‘reverse’ strand, though this is more a convention when dealing with paired-end data rather than an indication of the actual strand orientation.
AgendaIn this tutorial, we will cover:
Understanding Barcodes
Cell barcodes are designed primarily for delineating one cell from another, such that read transcripts containing different cell barcodes can be trivially said to be derived from different cells.
 Open image in new tab
Open image in new tabTranscript barcodes, meanwhile, are random sets of nucleotides added to each transcript.
 Open image in new tab
Open image in new tabThere are two things to take note of:
- The number of duplicates in the transcript barcodes (left)
- The number of duplicate read transcripts (right)
Transcript barcodes are often not unique. This becomes evident when you consider that there are approximately 200,000 mRNA’s in a given mammalian cell (Shapiro et al. 2013) which would require barcode lengths of greater than 9 nucleotides to capture, assuming no sequencing errors.
Question
- Why is it important to know which cell a read came from?
- Why do we need to barcode a read transcript too? Isn’t mapping it against the reference genome enough?
- If all our reads encode for a Red Gene (as above), we may want to know which cells express Red Gene more than others.
- e.g. If our Grey cell has 10 times more Red Gene reads than our Green cell, then we know that the Grey cell and Green cell differ in their expression of Red Gene - which might be biologically significant.
- Yes and no!
- Yes: We can indeed align our sequence against a reference genome and obtain the name of the gene it aligns against. This sequence will then contribute to the ‘count’ of sequences that gene has, and increase the expression of that gene.
- No: We do not know whether these ‘counts’ are unique. Many of these counts could be duplicates as a result of the amplification process. To explain further, we must look at UMIs and their role in the analysis.
The purpose of transcript barcodes is to reduce the impact of duplicated reads that occur non-linearly during the amplification process.
For this reason, transcript barcodes do not need to be unique. As long as we know that a given read maps to a specific transcript (i.e. after mapping it to a transcriptome), then we can assess how unique that read is based on:
- Cell barcode
- Transcript barcode
- Mapped location
To fully explore the uniqueness of counts, we must discuss the inclusion of UMIs in a single-cell analysis.
Mitigating duplicate transcript counts with UMIs
One of the major issues with sequencing is that the read fragments require amplification before they can be sequenced. A gene with a single mRNA transcript will not be detected by most sequencers, so it needs to be duplicated 100-1000x times for the sequencer to ‘see’ it.
Amplification is an imprecise process however, since some reads are amplified more than others, and subsequent amplification can lead to these over-amplified reads being over-amplified even more, leading to an exponential bias of some reads over others.
Note: Cell barcodes are not shown in any of the below examples, we assume they were added to our transcripts previously.
Naive Amplification
 Open image in new tab
Open image in new tabConsider the above example where two reads from different transcripts are amplified unevenly. The resulting frequency table would yield:
Reads in Cell 1 Gene Red 4 Gene Blue 0
But the truth is entirely different (i.e. Gene Red should have 1 count, and Gene Blue should also have 1 count). How do we correct for this bias?
Amplification with UMIs
Unique Molecular Identifiers (or UMIs) constitute the second portion of a barcode, where their role is to uniquely count reads such that amplicons of the same read are only counted once, e.g:
 Open image in new tab
Open image in new tabHere, we see two unique transcripts from Gene Red and two unique transcripts from Gene Blue, each given a (coloured) UMI. After amplification, Gene Red has more reads than Gene Blue. If we were to construct a frequency table as before to count the reads, we would have:
Reads in Cell 1 Gene Red 6 Gene Blue 3
This information is false, because it shows that Red has twice the expression that Blue does. However, we can reconstitute the true count by considering the UMI information:
UMI colour Reads in Cell 1 Gene Red Pink 2 Blue 4 Gene Blue Pink 1 Green 2
From this we can then make the decision to ignore the frequencies of these UMIs, and simply count how many unique UMIs we see in each gene:
Set of UMIs in Gene UMIs in Cell 1 Gene Red {Pink, Blue} 2 Gene Blue {Pink, Green} 2
This then provides us with the true count of the number of true transcripts for each gene as given by our original figure.
UMIs in Cell 1 Gene Red 2 Gene Blue 2
Question: about UMIs
- Are UMIs not specific to certain genes? Can the same UMI map to different genes?
- Can the same UMI map to different mRNA molecules of the same gene?
- Yes, UMIs are not specific to genes and the same UMI barcode can tag the transcripts of different genes. UMIs are not universal tags, they are just ‘added randomness’ that help reduce amplification bias.
- Yes, UMIs are not precise but operate probabilistically. In most cases, two transcripts of the same gene will be tagged by different UMIs. In rarer (but still prevalent) cases, the same UMI will capture different transcripts of the same gene.
- One helpful way to think about how quantification is performed is to observe the following hierarchy of data
Cell Barcode → Gene → UMIe.g.
BC:Cell Maps to Gene BC:UMI AAAT Slx1 TCA AAAT Slx2 GTG AAAT Gh13 TCA TTAA Slx1 TCA TTAA Atp3 CCC If UMIs were unique to a gene, then the
TCAUMI barcode would not have reads that map to both Slx1 and Gh13 in the same cell (AAAT).
Barcoding Format
We now know the role of UMIs and cell barcodes, but how do we handle them in the analysis? Let us look at 4 example sequences in our paired-end FASTQ data.
Hands On: Preparing the Data
- Create a new history and rename it (e.g. ‘Inspecting FastQ Files in scRNA batch data’)
- Import the following files from
Zenodoor from the data library (ask your instructor)https://zenodo.org/record/2573177/files/test_barcodes_celseq2_R1.fastq.gz https://zenodo.org/record/2573177/files/test_barcodes_celseq2_R2.fastq.gz
- Copy the link location
Click galaxy-upload Upload at the top of the activity panel
- Select galaxy-wf-edit Paste/Fetch Data
Paste the link(s) into the text field
Press Start
- Close the window
- Build a Dataset pair for the two FASTQ files
- Click on galaxy-selector “Select Items” at the top of the history panel
- Check the two boxes next to the R1 and R2 scRNA FASTQ samples
- Click 2 of N selected and choose Build dataset pair
- Ensure that the forward read is the
R1sample, and the reverse read is theR2sample.
- Click ‘Swap’ otherwise.
- Set the name of the pair
- Generate a list of reads to filter by creating a plain tabular file containing the following read names:
J00182:75:HTKJNBBXX:2:1114:12469:11073 J00182:75:HTKJNBBXX:2:2222:13301:35690 J00182:75:HTKJNBBXX:2:1203:25022:13763 J00182:75:HTKJNBBXX:2:1115:8501:46961- Set the datatype of the file as tabular

At this point we now have a history with two items: our paired FASTQ test data, and a tabular file of read names. We will now apply the tabular file to the FASTQ file and extract only those reads.
Hands On: Extracting the Reads
- Extracting our 4 reads
- Filter sequences by ID ( Galaxy version 0.2.9) with the following parameters:
- Sequence file to be filtered
- Click the Dataset Collection icon
- Select the FastQ collection if not already selected.
- Filter using the ID list from:
tabular file
- Tabular file containing sequence identifiers:
Pasted Entry- Column(s) containing sequence identifiers
- Select/Unselect all:(tick the box)
- Output positive matches, negative matches, or both?:
Just positive matches (ID on list), as a single fileChange the datatypes of the output pair to
fastqsangerif not already set.
- Click on the galaxy-pencil pencil icon for the dataset to edit its attributes
- In the central panel, click galaxy-chart-select-data Datatypes tab on the top
- In the galaxy-chart-select-data Assign Datatype, select your desired datatype from “New Type” dropdown
- Tip: you can start typing the datatype into the field to filter the dropdown menu
- Click the Save button
- Viewing our 4 reads side-by-side
- Activate the Window Manager by clicking on the Enable/Disable Window Manager icon on the main top toolbar
- Click on the newly generated FastQ pair ending in “with matched ID” to expand the individual reads
- Click on the galaxy-eye symbol of the forward read
- Click somewhere outside the white box to close the Window Manager
- Click on the galaxy-eye symbol of the reverse read
- Position/Resize the boxes as desired
If you would like to view two or more datasets at once, you can use the Window Manager feature in Galaxy:
- Click on the Window Manager icon galaxy-scratchbook on the top menu bar.
- You should see a little checkmark on the icon now
- View galaxy-eye a dataset by clicking on the eye icon galaxy-eye to view the output
- You should see the output in a window overlayed over Galaxy
- You can resize this window by dragging the bottom-right corner
- View galaxy-eye a second dataset from your history
- You should now see a second window with the new dataset
- This makes it easier to compare the two outputs
- Repeat this for as many files as you would like to compare
- You can turn off the Window Manager galaxy-scratchbook by clicking on the icon again
Our Four Reads of Interest
Let us examine these four reads of interest which we have just sub-selected using their headers:
@J00182:75:HTKJNBBXX:2:1115:8501:46961 1:N:0:ATCACG GGAAGAACCAGATTTTTTTTTTTTTTTTTT + AAFFFJJJJJJJFFFJJJJJJJJJJJJJJJ @J00182:75:HTKJNBBXX:2:1203:25022:13763 1:N:0:ATCACG GTCCCAGGTAACTTTTTTTTTTTTTTTTTT + AAFFFJJJJJJJJFFJJJJJJJJJFJ<FF- @J00182:75:HTKJNBBXX:2:2222:13301:35690 1:N:0:ATCACG GTCCCAGGTAACTTTTTTTTTTTTTTTTTT + AAFFFJJJJJJJ<AFJJJJJFFJJFJJJFF @J00182:75:HTKJNBBXX:2:1114:12469:11073 1:N:0:ATCACG CGGCGTGGTAACTTTTTTTTTTTTTTTTCC + AAFFFJJJJJJJFAFFJJJJJJJJF---<F
@J00182:75:HTKJNBBXX:2:1115:8501:46961 2:N:0:ATCACG GACCTCTGATCTTTACGAAAGGCCAACGCGTTTTCAGTCTGGACACGGTTCAGCTCCTGTTCATTATTCA + A<<A-777F<AA<AJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJ @J00182:75:HTKJNBBXX:2:1203:25022:13763 2:N:0:ATCACG GCCACCTAATTTCCGTCATCACACTCCTCTCCGTTTTCAACTTGCACAATGCTGTCTCCGCAGAATCCCT + ---<----<A---77-7A-FJ<JJFFJJ<JJAJ7<-FAFFJJFF<FFJJFFAJFA-AFFFJFFFFFJAJJ @J00182:75:HTKJNBBXX:2:2222:13301:35690 2:N:0:ATCACG CAATCCTCTCCGTTATCAACTTGCACAATGCTGTCTCCGCAGAATCCCTCCGGATCAGGATCGCTCTCCA + <<A-77--77F<----7AFJ-A--FJJJFAJF-AFAJAJ<JFJ<JJJFFJJJFJJJJJAAFJJJFJJJF- @J00182:75:HTKJNBBXX:2:1114:12469:11073 2:N:0:ATCACG ATCCACTTATTGCAAAGCAGAGGACATTGAGTCTCACCTTTTGTCCAGGTCTTCCAATTTCACCCTGCAA + A-77AA-7FF<7FFJFFFJJJJJJJJJJJJJ-AFJJJJJJJFJJJJJJJJJJJJJJJJJJJJJJJJJJJJ
What we observe are the standard four lines of any FASTQ file:
- Read name starting with
@ - Sequence of nucleotide bases
- Separator
+ - Quality string of the nucleotide bases in ASCII
The main source of interest for us is in the (2) sequences of these reads, which somewhere within encode for three crucial pieces of information that we will need to perform quantification:
- Cell Barcode
- UMI Barcode
- Reverse-transcribed mRNA sequence
These can be encoded into the sequences of our paired-end data by any means. In order to know where our barcodes are, we must be familiar with the sequencing primers used in the analysis:
Verifying the Barcode Format
As shown in CEL-Seq2 protocol, we have the following encoding:
- Forward Read:
- 01-06bp: UMI Barcode
- 07-12bp: Cell Barcode
- 13-30bp: Poly-T tail
- Reverse Read:
- 01-70bp: mRNA sequence
The encoding of the barcodes on the first read can actually be seen by examining the distribution of bases in a FastQC plot.
Hands On: Confirming the Barcoding
- FastQC ( Galaxy version 0.74+galaxy1) with the following parameters:
- param-collection “Short read data from your current history”:
Paired FastQ(the original paired set) You will need to choose ‘Dataset collection’ to allow this as an input.CommentWe are only interested in the distribution of bases on the Forward read, but it is more convenient to process the data as a pair instead of un-hiding the original dataset
- Click on FastQC on collection :Webpage
- Click on the galaxy-eye of the Forward read
- Click on the Per base sequence content header on the side-bar
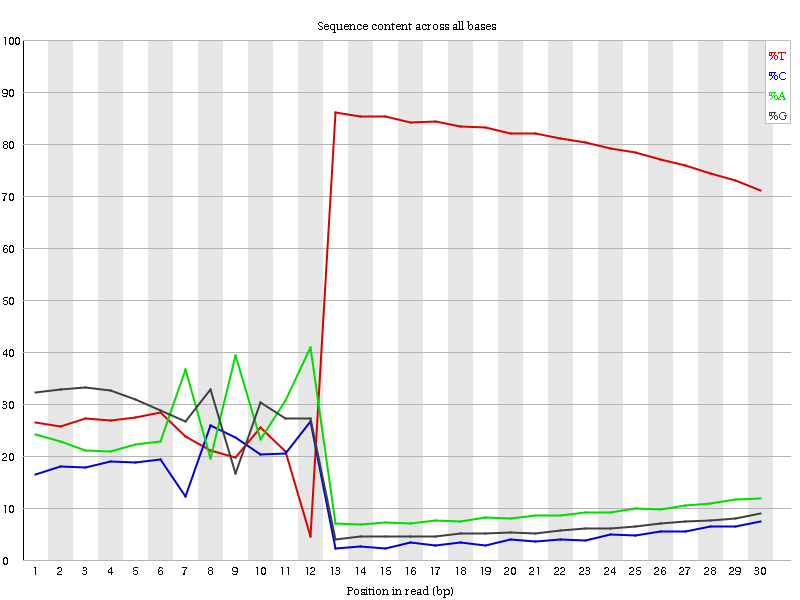 Open image in new tab
Open image in new tabHere we can see the three distinct regions along the x-axis that correspond to our expected CEL-Seq2 Schema:
- 01-06bp: smooth, relatively constant bases.
- 07-12bp: noisy, highly varied distribution of bases.
- 13-30bp: T-dominated region
We can see that the distribution of nucleotides in the 01-06bp range is relatively more stable than the distribution of nucleotides in the 07-12bp range, which seems to exhibit more extreme variation.
QuestionWhy is this the case? Why is the UMI barcode base distribution smoother than the Cell barcode base distribution?
There are far more UMIs than cells. Cell barcodes are designed and selected with a specified edit distance, greatly limiting their availability in the data. UMIs are not so well-curated – i.e it is possible to encounter the same UMI in the same cell multiple times. The more extreme variation in the 7-12bp region is simply caused by a fewer number of samples.
Uniting Barcodes with Sequence
In a sense, we have a disparity in our data: the reverse reads contain the sequences we wish to map, but not the barcodes; the forward reads contain the barcode, but not the sequence. For the forward and reverse reads given above, the information that we really want from both can be summarized in this table:
| Read | Cell | UMI | Sequence |
|---|---|---|---|
| @J00182:75:HTKJNBBXX:2:1115:8501:46961 | ACCAGA | GGAAGA | GACCTCTGATCTTTACGAAAGGCCAACGCGTTTTCAGTCTGGACACGGTTCAGCTCCTGTTCATTATTCA |
| @J00182:75:HTKJNBBXX:2:1203:25022:13763 | GGTAAC | GTCCCA | GCCACCTAATTTCCGTCATCACACTCCTCTCCGTTTTCAACTTGCACAATGCTGTCTCCGCAGAATCCCT |
| @J00182:75:HTKJNBBXX:2:2222:13301:35690 | GGTAAC | GTCCCA | CAATCCTCTCCGTTATCAACTTGCACAATGCTGTCTCCGCAGAATCCCTCCGGATCAGGATCGCTCTCCA |
| @J00182:75:HTKJNBBXX:2:1114:12469:11073 | GGTAAC | CGGCGT | ATCCACTTATTGCAAAGCAGAGGACATTGAGTCTCACCTTTTGTCCAGGTCTTCCAATTTCACCCTGCAA |
QuestionProvided that these reads all map to the same gene:
- Which of these reads come from the same cell?
- Which of these reads are PCR duplicates?
- Reads:
@J00182:75:HTKJNBBXX:2:1203:25022:13763@J00182:75:HTKJNBBXX:2:2222:13301:35690@J00182:75:HTKJNBBXX:2:1114:12469:11073all have the cell barcodeGGTAAC.- Reads:
@J00182:75:HTKJNBBXX:2:1203:25022:13763@J00182:75:HTKJNBBXX:2:2222:13301:35690appear to be PCR duplicates, since they both have the same cell barcode and same UMI.However if we consider their sequences, we can see that they contain different (but overlapping) sequences.
13763: GCCACCTAATTTCCGTCATCACACTCCTCTCCGTTTTCAACTTGCACAATGCTGTCTCCGCAGAATCCCT 35690: CAATCCTCTCCGTTATCAACTTGCACAATGCTGTCTCCGCAGAATCCCTCCGGATCAGGATCGCTCTCCAThey describe the same transcript but have sequences from different reads, and therefore both reads should be counted as separate reads. Whether or not both these reads are counted as a single read due to their identical barcodes, or counted separately due to their differing sequences depends entirely on the deduplication utility they are fed into it.
Coupling our Data Sources
How should we unite these two source of information into a single location without impacting the data content?
For this we need to take the barcode information from the Forward reads, and stick it into the header of the Reverse reads. That way we can align our sequence to the reference and still keep the barcode information associated with the reads.
Hands On: Barcode Extraction and Annotation of our 4 reads
- UMI-tools extract ( Galaxy version 1.1.6+galaxy0) with the following parameters:
- “Library type”:
Paired-end Dataset Collection
- param-collection “Reads in FASTQ format”:
output(Our paired set of 4 sequences)- “Barcode on both reads?”:
Barcode on first read only- “Barcode pattern for first read”:
NNNNNNCCCCCC- “Enable quality filter?”:
NoComment
- Here we specify the format of our barcodes as
NNNNNNCCCCCCwhere the Ns represent UMI bases and the Cs represent the cell barcodes.- In some protocols, actual sequence data can be found in between the cell and UMI barcodes, wherein it is neccesary to represent sequence bases using X.
- e.g. A protocol that starts with a 3bp sequence, followed by a 4bp Cell barcode, followed once again by a 10bp sequence, and then finally a 5bp UMI barcode, would require the following barcode format:
XXXCCCCXXXXXXXXXXNNNNN- Click the galaxy-eye symbol on the Reads1: UMI-tools extract file
- Click somewhere outside the white box to close the Window Manager
- Click the galaxy-eye symbol on the Reads2: UMI-tools extract file
We should now be able to see the following reads:
@J00182:75:HTKJNBBXX:2:1115:8501:46961_ACCAGA_GGAAGA 1:N:0:ATCACG TTTTTTTTTTTTTTTTTT + FFFJJJJJJJJJJJJJJJ @J00182:75:HTKJNBBXX:2:1203:25022:13763_GGTAAC_GTCCCA 1:N:0:ATCACG TTTTTTTTTTTTTTTTTT + JFFJJJJJJJJJFJ<FF- @J00182:75:HTKJNBBXX:2:2222:13301:35690_GGTAAC_GTCCCA 1:N:0:ATCACG TTTTTTTTTTTTTTTTTT + <AFJJJJJFFJJFJJJFF @J00182:75:HTKJNBBXX:2:1114:12469:11073_GGTAAC_CGGCGT 1:N:0:ATCACG TTTTTTTTTTTTTTTTCC + FAFFJJJJJJJJF---<F>
@J00182:75:HTKJNBBXX:2:1115:8501:46961_ACCAGA_GGAAGA 2:N:0:ATCACG GACCTCTGATCTTTACGAAAGGCCAACGCGTTTTCAGTCTGGACACGGTTCAGCTCCTGTTCATTATTCA + A<<A-777F<AA<AJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJ @J00182:75:HTKJNBBXX:2:1203:25022:13763_GGTAAC_GTCCCA 2:N:0:ATCACG GCCACCTAATTTCCGTCATCACACTCCTCTCCGTTTTCAACTTGCACAATGCTGTCTCCGCAGAATCCCT + ---<----<A---77-7A-FJ<JJFFJJ<JJAJ7<-FAFFJJFF<FFJJFFAJFA-AFFFJFFFFFJAJJ @J00182:75:HTKJNBBXX:2:2222:13301:35690_GGTAAC_GTCCCA 2:N:0:ATCACG CAATCCTCTCCGTTATCAACTTGCACAATGCTGTCTCCGCAGAATCCCTCCGGATCAGGATCGCTCTCCA + <<A-77--77F<----7AFJ-A--FJJJFAJF-AFAJAJ<JFJ<JJJFFJJJFJJJJJAAFJJJFJJJF- @J00182:75:HTKJNBBXX:2:1114:12469:11073_GGTAAC_CGGCGT 2:N:0:ATCACG ATCCACTTATTGCAAAGCAGAGGACATTGAGTCTCACCTTTTGTCCAGGTCTTCCAATTTCACCCTGCAA + A-77AA-7FF<7FFJFFFJJJJJJJJJJJJJ-AFJJJJJJJFJJJJJJJJJJJJJJJJJJJJJJJJJJJJ
Notice the remaining sequence in each of the reads, and that the reverse reads appear to fully encapsulate all the information that we wanted to capture in our table at the beginning of this section.
Question
- Compare the Forward/Read1 and Reverse/Read2 reads to those prior the extraction. What has happened to the header and sequence of each read?
- Are the Forward reads useful at all?
- Comparison:
- Forward:
- Sequence: The
cellandumisections of the sequence have been removed, leaving behind only the PolyT tail.- Header: The
cellandumisections of the sequence have been added ascell_umibarcode in the header- Reverse:
- Sequence: Has not changed.
- Header: The
cellandumisections of the sequence from the Forward (note: NOT Reverse) reads have been added to the header.- With the inclusion of the cell and UMI barcodes into the header of our sequence data, we now have all our useful data in the Reverse reads. We can now effectively throw away our Forward reads, as they have no more useful information within them.
We have now successfully multiplexed data from several different (cell) sources into a single file that will greatly simplify the mapping/alignment process downstream.
We have also now successfully de-multiplexed our data, by decoding each pair of reads into barcoding and sequence parts and making use of the FASTQ format by storing this information within the FASTQ headers and data, respectively.
Conclusion
With this tutorial we have understood the importance of handling FASTQ data from different sources, and extracting the information we need (barcodes (cell and UMI) and sequence) using UMI-tools so that we can perform mapping without losing any context of where the reads are derived from.
This tutorial is part of the https://singlecell.usegalaxy.eu portal (Tekman et al. 2020).
You've Finished the Tutorial
Key points
Verifying the distribution of barcodes via a FASTQC plot
Relocating barcodes into headers
Removing unwanted barcodes
Frequently Asked Questions
Have questions about this tutorial? Have a look at the available FAQ pages and support channelsUseful literature
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
References
- Shapiro, E., T. Biezuner, and S. Linnarsson, 2013 Single-cell sequencing-based technologies will revolutionize whole-organism science. Nature Reviews Genetics 14: 618–630. 10.1038/nrg3542
- Hashimshony, T., N. Senderovich, G. Avital, A. Klochendler, Y. de Leeuw et al., 2016 CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biology 17: 10.1186/s13059-016-0938-8
- Tekman, M., B. Batut, A. Ostrovsky, C. Antoniewski, D. Clements et al., 2020 A single-cell RNA-sequencing training and analysis suite using the Galaxy framework. GigaScience 9: giaa102. 10.1093/gigascience/giaa102 https://academic.oup.com/gigascience/article/9/10/giaa102/5931798
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.

Citing this Tutorial
- Mehmet Tekman, Understanding Barcodes (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/single-cell/tutorials/scrna-umis/tutorial.html Online; accessed TODAY
- Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
Congratulations on successfully completing this tutorial!@misc{single-cell-scrna-umis, author = "Mehmet Tekman", title = "Understanding Barcodes (Galaxy Training Materials)", year = "", month = "", day = "", url = "\url{https://training.galaxyproject.org/training-material/topics/single-cell/tutorials/scrna-umis/tutorial.html}", note = "[Online; accessed TODAY]" } @article{Hiltemann_2023, doi = {10.1371/journal.pcbi.1010752}, url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752}, year = 2023, month = {jan}, publisher = {Public Library of Science ({PLoS})}, volume = {19}, number = {1}, pages = {e1010752}, author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and}, editor = {Francis Ouellette}, title = {Galaxy Training: A powerful framework for teaching!}, journal = {PLoS Comput Biol} }
Do you want to extend your knowledge?Follow one of our recommended follow-up trainings:
You can use Ephemeris's
shed-tools installcommand to install the tools used in this tutorial.shed-tools install [-g GALAXY] [-a API_KEY] -t <(curl https://training.galaxyproject.org/training-material/api/topics/single-cell/tutorials/scrna-umis/tutorial.json | jq .admin_install_yaml -r)Alternatively you can copy and paste the following YAML
--- install_tool_dependencies: true install_repository_dependencies: true install_resolver_dependencies: true tools: - name: fastqc owner: devteam revisions: 2c64fded1286 tool_panel_section_label: FASTA/FASTQ tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: umi_tools_extract owner: iuc revisions: 158e9e91b9fc tool_panel_section_label: FASTA/FASTQ tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: seq_filter_by_id owner: peterjc revisions: 85ef5f5a0562 tool_panel_section_label: Filter and Sort tool_shed_url: https://toolshed.g2.bx.psu.edu/