Checking expected species and contamination in bacterial isolate
| Author(s) |
|
| Editor(s) |
|
| Reviewers |
|
OverviewQuestions:
Objectives:
What are the species in bacterial isolate sequencing data?
Requirements:
Run a series of tool to identify species in bacterial isolate sequencing data
Visualize the species abundance
Time estimation: 1 hourLevel: Introductory IntroductorySupporting Materials:Published: Mar 4, 2024Last modification: Apr 8, 2026License: Tutorial Content is licensed under Creative Commons Attribution 4.0 International License. The GTN Framework is licensed under MITpurl PURL: https://gxy.io/GTN:T00423rating Rating: 4.0 (0 recent ratings, 2 all time)version Revision: 6
Sequencing (determining of DNA/RNA nucleotide sequence) is used all over the world for all kinds of analysis. The product of these sequencers are reads, which are sequences of detected nucleotides.
When sequencing bacterial isolates, it might be good to check if the expected species or strains can be identified in the data or if there is any contamination.
To illustrate the process, we take quality-controlled data of a bacterial genome (KUN1163 sample) from sequencing data produced in “Complete Genome Sequences of Eight Methicillin-Resistant Staphylococcus aureus Strains Isolated from Patients in Japan” (Hikichi et al. 2019).
Methicillin-resistant Staphylococcus aureus (MRSA) is a major pathogen causing nosocomial infections, and the clinical manifestations of MRSA range from asymptomatic colonization of the nasal mucosa to soft tissue infection to fulminant invasive disease. Here, we report the complete genome sequences of eight MRSA strains isolated from patients in Japan.
AgendaIn this tutorial, we will cover:
Galaxy and data preparation
Any analysis should get its own Galaxy history. So let’s start by creating a new one and get the data (forward and reverse quality-controlled sequences) into it.
Hands On: Prepare Galaxy and data
Create a new history for this analysis
To create a new history simply click the new-history icon at the top of the history panel:
Rename the history
- Click on galaxy-pencil (Edit) next to the history name (which by default is “Unnamed history”)
- Type the new name
- Click on Save
- To cancel renaming, click the galaxy-undo “Cancel” button
If you do not have the galaxy-pencil (Edit) next to the history name (which can be the case if you are using an older version of Galaxy) do the following:
- Click on Unnamed history (or the current name of the history) (Click to rename history) at the top of your history panel
- Type the new name
- Press Enter
Import the quality-controlled sequences from Zenodo or from Galaxy shared data libraries:
https://zenodo.org/record/10572227/files/DRR187559_after_fastp_1.fastq.gz https://zenodo.org/record/10572227/files/DRR187559_after_fastp_2.fastq.gz
- Copy the link location
Click galaxy-upload Upload at the top of the activity panel
- Select galaxy-wf-edit Paste/Fetch Data
Paste the link(s) into the text field
Press Start
- Close the window
As an alternative to uploading the data from a URL or your computer, the files may also have been made available from a shared data library:
- Go into Libraries (left panel)
- Navigate to the correct folder as indicated by your instructor.
- On most Galaxies tutorial data will be provided in a folder named GTN - Material –> Topic Name -> Tutorial Name.
- Select the desired files
- Click on Add to History galaxy-dropdown near the top and select as Datasets from the dropdown menu
In the pop-up window, choose
- “Select history”: the history you want to import the data to (or create a new one)
- Click on Import
Create a paired collection named
Paired Reads
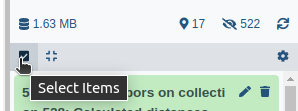
- Click on galaxy-selector Select Items at the top of the history panel
- Check all the datasets in your history you would like to include
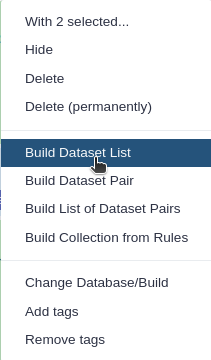
Click n of N selected and choose Advanced Build List
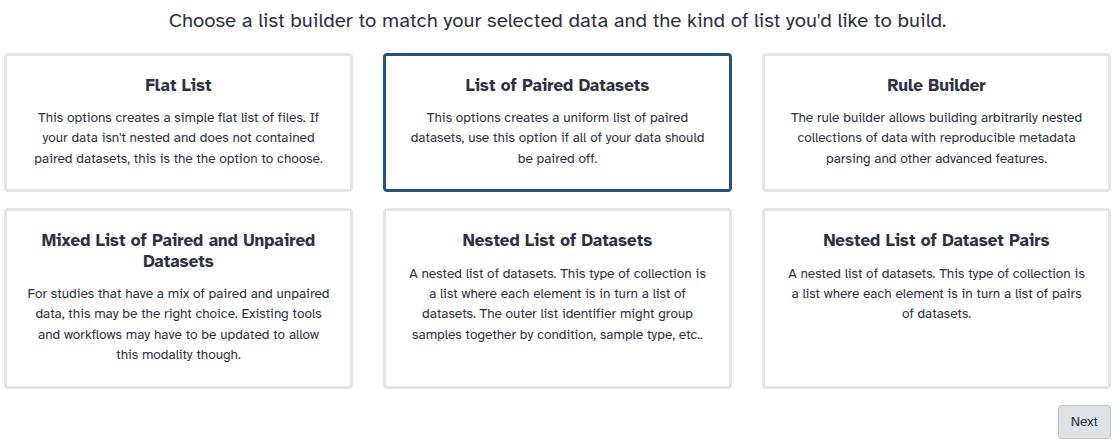
You are in the collection building wizard. Choose List of Paired Datasets and click ‘Next’ button at the right bottom corner.
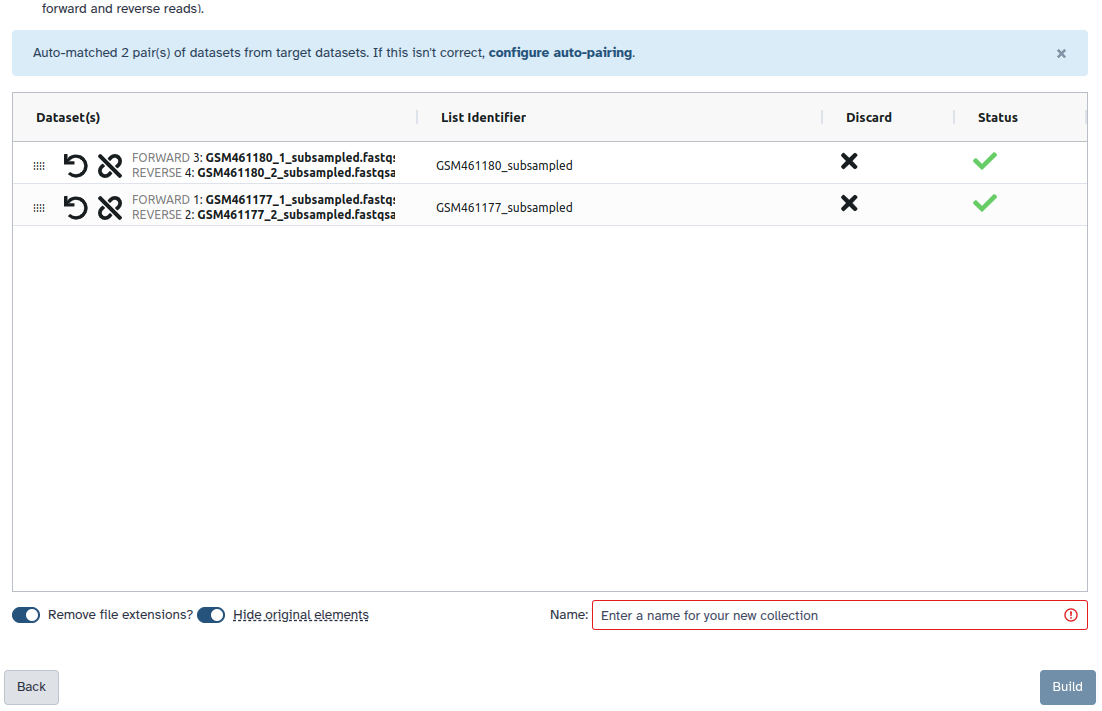
Check and configure auto-pairing. Commonly matepairs have suffix
_1and_2or_R1and_R2. Click on ‘Next’ at the bottom.
- Edit the List Identifier as required.
- Enter a name for your collection
- Click Build to build your collection
- Click on the checkmark icon at the top of your history again


Taxonomic profiling
To find out which microorganisms are present, we will compare the reads of the sample to a reference database, i.e. sequences of known microorganisms stored in a database, using Kraken2 (Wood et al. 2019).
In the \(k\)-mer approach for taxonomy classification, we use a database containing DNA sequences of genomes whose taxonomy we already know. On a computer, the genome sequences are broken into short pieces of length \(k\) (called \(k\)-mers), usually 30bp.
Kraken examines the \(k\)-mers within the query sequence, searches for them in the database, looks for where these are placed within the taxonomy tree inside the database, makes the classification with the most probable position, then maps \(k\)-mers to the lowest common ancestor (LCA) of all genomes known to contain the given \(k\)-mer.
Kraken2 uses a compact hash table, a probabilistic data structure that allows for faster queries and lower memory requirements. It applies a spaced seed mask of s spaces to the minimizer and calculates a compact hash code, which is then used as a search query in its compact hash table; the lowest common ancestor (LCA) taxon associated with the compact hash code is then assigned to the k-mer.
You can find more information about the Kraken2 algorithm in the paper Improved metagenomic analysis with Kraken 2.
 of the genomes that contain that k-mer in the database. The taxa associated with the sequence's k-mers, as well as the taxa's ancestors, form a pruned subtree of the general taxonomy tree, which is used for classification. In the classification tree, each node has a weight equal to the number of k-mers in the sequence associated with the node's taxon. Each root-to-leaf (RTL) path in the classification tree is scored by adding all weights in the path, and the maximal RTL path in the classification tree is the classification path (nodes highlighted in yellow). The leaf of this classification path (the orange, leftmost leaf in the classification tree) is the classification used for the query sequence. Source: <span class=\"citation\"><a href=\"#Wood2014\">Wood and Salzberg 2014</a></span>")
For this tutorial, we will use the PlusPF database which contains the Standard (archaea, bacteria, viral, plasmid, human, UniVec_Core), protozoa and fungi data.
Hands On: Assign taxonomic labels with Kraken2
- Kraken2 ( Galaxy version 2.1.3+galaxy2) with the following parameters:
- “Single or paired reads”:
Paired
- param-collection “Collection of paired reads”:
Paired Reads- “Minimum Base Quality”:
10- In “Create Report”:
- “Print a report with aggregrate counts/clade to file”:
Yes- “Select a Kraken2 database”:
PlusPF-16
-
Classification: tabular files with one line for each sequence classified by Kraken and 5 columns:
C/U: a one letter indicating if the sequence classified or unclassified- Sequence ID as in the input file
- NCBI taxonomy ID assigned to the sequence, or 0 if unclassified
- Length of sequence in bp (
read1|read2for paired reads) -
A space-delimited list indicating the lowest common ancestor (LCA) mapping of each k-mer in the sequence
For example,
562:13 561:4 A:31 0:1 562:3would indicate that:- The first 13 k-mers mapped to taxonomy ID #562
- The next 4 k-mers mapped to taxonomy ID #561
- The next 31 k-mers contained an ambiguous nucleotide
- The next k-mer was not in the database
- The last 3 k-mers mapped to taxonomy ID #562
|:|indicates end of first read, start of second read for paired reads
Column 1 Column 2 Column 3 Column 4 Column 5 C DRR187559.1 1280 164|85 0:1 1279:1 0:41 1279:10 0:5 1280:5 1279:1 0:1 1279:1 0:7 1279:5 0:2 1279:6 0:12 1279:5 0:19 1279:2 0:6 |:| 0:39 1279:2 0:6 1279:3 0:1 C DRR187559.2 1280 70|198 0:2 1279:5 0:29 |:| 0:52 1279:5 0:13 1279:3 0:23 1279:2 0:45 1280:1 0:9 1280:3 A:8 C DRR187559.3 1279 106|73 0:4 1279:3 0:36 1279:4 0:10 1279:5 0:3 1279:5 0:2 |:| 0:39 C DRR187559.4 1279 121|189 1279:6 0:17 1279:4 0:28 1279:2 0:30 |:| 0:7 1279:5 0:19 1279:5 0:20 1279:1 0:8 1279:1 0:1 1279:6 0:25 1279:2 0:44 A:11 C DRR187559.5 1279 68|150 1279:2 0:20 1279:3 0:9 |:| 0:10 1279:3 0:24 1279:2 0:9 1279:5 0:21 1279:5 0:20 1279:5 0:9 1279:1 0:2 C DRR187559.6 1280 137|246 0:2 1280:5 0:28 1279:1 0:28 1279:2 0:8 1279:2 0:23 1279:1 0:3 |:| 1279:1 0:2 1279:3 0:61 1279:2 0:14 1279:2 0:97 A:30Question- Is the first sequence in the file classified or unclassified?
- What is the taxonomy ID assigned to the first classified sequence?
- What is the corresponding taxon?
- classified
- 1280
- 1280 corresponds to Staphylococcus aureus .
-
Report: tabular files with one line per taxon and 6 columns or fields
- Percentage of fragments covered by the clade rooted at this taxon
- Number of fragments covered by the clade rooted at this taxon
- Number of fragments assigned directly to this taxon
- A rank code, indicating
- (U)nclassified
- (R)oot
- (D)omain
- (K)ingdom
- (P)hylum
- (C)lass
- (O)rder
- (F)amily
- (G)enus, or
- (S)pecies
Taxa that are not at any of these 10 ranks have a rank code that is formed by using the rank code of the closest ancestor rank with a number indicating the distance from that rank. E.g.,
G2is a rank code indicating a taxon is between genus and species and the grandparent taxon is at the genus rank. - NCBI taxonomic ID number
- Indented scientific name
Column 1 Column 2 Column 3 Column 4 Column 5 Column 6 0.24 1065 1065 U 0 unclassified 99.76 450716 14873 R 1 root 96.44 435695 2 R1 131567 cellular organisms 96.43 435675 3889 D 2 Bacteria 95.56 431709 78 D1 1783272 Terrabacteria group 95.53 431578 163 P 1239 Firmicutes 95.49 431390 4625 C 91061 Bacilli 94.38 426383 1436 O 1385 Bacillales 94.04 424874 2689 F 90964 Staphylococcaceae 93.44 422124 234829 G 1279 StaphylococcusQuestion- How many taxa have been found?
- What are the percentage on unclassified?
- What are the domains found?
- 621, as the number of lines
- 0.24%
- Only Bacteria
Species extraction
In Kraken output, there are quite a lot of identified taxa with different levels. To extract the species level, we will use Bracken.
Bracken (Bayesian Reestimation of Abundance after Classification with Kraken) is a “simple and worthwile addition to Kraken for better abundance estimates” (Ye et al. 2019). Instead of only using proportions of classified reads, it takes a probabilistic approach to generate final abundance profiles. It works by re-distributing reads in the taxonomic tree: “Reads assigned to nodes above the species level are distributed down to the species nodes, while reads assigned at the strain level are re-distributed upward to their parent species” (Lu et al. 2017).
Hands On: Extract species with Bracken
- Bracken ( Galaxy version 2.9+galaxy0) with the following parameters:
- param-collection “Kraken report file”: Report output of Kraken
“Select a kmer distribution”:
PlusPF, same as for KrakenIt is important to choose the same database that you also chose for Kraken2
- “Level”:
Species
Bracken generates one output as a table with 7 columns:
- Taxon name
- Taxonomy ID
- Level ID (S=Species, G=Genus, O=Order, F=Family, P=Phylum, K=Kingdom)
- Kraken assigned reads
- Added reads with abundance re-estimation
- Total reads after abundance re-estimation
- Fraction of total reads
Question
- How many species have been found?
- Which the species has been the most identified? And in which proportion?
- What are the other species?
- 51 (52 lines including 1 line with header)
- Staphylococcus aureus with 95.5% of the reads.
- Most of the other species are from Staphylococcus genus, so same as Staphylococcus aureus. The other species in really low proportion.
As expected Staphylococcus aureus represents most of the reads in the data.
Contamination identification
To explore Kraken report and specially to detect more reliably minority organisms or contamination, we will use Recentrifuge (Martı́ Jose Manuel 2019).
Hands On: Identify contamination
- Recentrifuge ( Galaxy version 1.12.1+galaxy0) with the following parameters:
- param-collection “Select taxonomy file tabular formated”: Classification output of Kraken2 tool
- “Type of input file (Centrifuge, CLARK, Generic, Kraken, LMAT)”:
Kraken- In “Database type”:
- “Cached database whith taxa ID”: latest
- In “Output options”:
- “Type of extra output to be generated (default on CSV)”:
TSV- “Remove the log file”:
Yes- In ” Fine tuning of algorithm parameters”:
- “Strain level instead of species as the resolution limit for the robust contamination removal algorithm; use with caution, this is an experimental feature”:
Yes
Recentrifuge generates 3 outputs:
-
A statistic table with general statistics about assignations
Question- How many sequences have been used?
- How many sequences have been classified?
- 451,780
- 450,715
-
A data table with a report for each taxa
Question- How many taxa have been kept?
- What is the lowest level in the data?
- 185 (189 lines including 3 header lines)
- The lowest level is strain.
-
A HTML report with a Krona chart
Question- What is the percentage of classified sequences?
- When clicking on Staphylococcus aureus, what can we say about the strains?
- Is there any contamination?
- 99.8%
- 99% of sequences assigned to Staphylococcus aureus are not assigned to any strains, probably because they are too similar to several strains. Staphylococcus aureus subsp. aureus JKD6159 is the strain with the most classified sequences, but only 0.3% of the sequences assigned to Staphylococcus aureus.
- There is no sign of a possible contamination. Most sequences are classified to taxon on the Staphylococcus aureus taxonomy. Only 3% of the sequences are not classified to Staphylococcus.
Conclusion
In this tutorial, we checked bacterial isolate sequencing data for expected species and potential contamination.
You've Finished the Tutorial
Key points
Kraken assigns taxons to sequences
Bracken extracts species from Kraken assignations
Recentrifuge extracts stats and creates visualization from Kraken report
Frequently Asked Questions
Have questions about this tutorial? Have a look at the available FAQ pages and support channelsUseful literature
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
References
- Wood, D. E., and S. L. Salzberg, 2014 Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biology 15: R46. 10.1186/gb-2014-15-3-r46
- Lu, J., F. P. Breitwieser, P. Thielen, and S. L. Salzberg, 2017 Bracken: estimating species abundance in metagenomics data. PeerJ Computer Science 3: e104. 10.7717/peerj-cs.104
- Hikichi, M., M. Nagao, K. Murase, C. Aikawa, T. Nozawa et al., 2019 Complete Genome Sequences of Eight Methicillin-Resistant Staphylococcus aureus Strains Isolated from Patients in Japan (I. L. G. Newton, Ed.). Microbiology Resource Announcements 8: 10.1128/mra.01212-19
- Martı́ Jose Manuel, 2019 Recentrifuge: Robust comparative analysis and contamination removal for metagenomics. PLoS computational biology 15: e1006967. 10.1371/journal.pcbi.100696
- Wood, D. E., J. Lu, and B. Langmead, 2019 Improved metagenomic analysis with Kraken 2. Genome biology 20: 1–13. 10.1186/s13059-019-1891-0
- Ye, S. H., K. J. Siddle, D. J. Park, and P. C. Sabeti, 2019 Benchmarking Metagenomics Tools for Taxonomic Classification. Cell 178: 779–794. 10.1016/j.cell.2019.07.010
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.

Citing this Tutorial
- Bérénice Batut, Checking expected species and contamination in bacterial isolate (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/ecology/tutorials/bacterial-isolate-species-contamination-checking/tutorial.html Online; accessed TODAY
- Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{ecology-bacterial-isolate-species-contamination-checking, author = "Bérénice Batut", title = "Checking expected species and contamination in bacterial isolate (Galaxy Training Materials)", year = "", month = "", day = "", url = "\url{https://training.galaxyproject.org/training-material/topics/ecology/tutorials/bacterial-isolate-species-contamination-checking/tutorial.html}", note = "[Online; accessed TODAY]" } @article{Hiltemann_2023, doi = {10.1371/journal.pcbi.1010752}, url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752}, year = 2023, month = {jan}, publisher = {Public Library of Science ({PLoS})}, volume = {19}, number = {1}, pages = {e1010752}, author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and}, editor = {Francis Ouellette}, title = {Galaxy Training: A powerful framework for teaching!}, journal = {PLoS Comput Biol} }
Funding
These individuals or organisations provided funding support for the development of this resource
Congratulations on successfully completing this tutorial!

You can use Ephemeris's
shed-tools installcommand to install the tools used in this tutorial.shed-tools install [-g GALAXY] [-a API_KEY] -t <(curl https://training.galaxyproject.org/training-material/api/topics/ecology/tutorials/bacterial-isolate-species-contamination-checking/tutorial.json | jq .admin_install_yaml -r)Alternatively you can copy and paste the following YAML
--- install_tool_dependencies: true install_repository_dependencies: true install_resolver_dependencies: true tools: - name: bracken owner: iuc revisions: 1d4bd12f01cf tool_panel_section_label: Metagenomic Analysis tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: bracken owner: iuc revisions: 4d350ac5148e tool_panel_section_label: Metagenomic Analysis tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: kraken2 owner: iuc revisions: e150ae930235 tool_panel_section_label: Metagenomic Analysis tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: recentrifuge owner: iuc revisions: b5407cc2bf51 tool_panel_section_label: Metagenomic Analysis tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: recentrifuge owner: iuc revisions: 613ee22af97c tool_panel_section_label: Metagenomic Analysis tool_shed_url: https://toolshed.g2.bx.psu.edu/