Query an annotated mobile genetic element database to identify and annotate genetic elements (e.g. plasmids) in metagenomics data
| Author(s) |
|
| Reviewers |
|
OverviewQuestions:
Objectives:
How can we use an existing database annotated database to identify and annotate genetic elements in metagenomics data?
Requirements:
Perform metagenomics read mapping against mobile genetic element database.
Evaluate the distribution of mapping scores to identify high-quality alignments.
Evaluate plasmid coverage to determine effective filtering thresholds.
Filter alignments based on plasmid coverage and mapping quality.
Justify the filtering thresholds chosen for identifying plasmid sequences.
Generate a curated table of plasmid sequences and convert it into a FASTA file for further analysis.
Use tools to process sequences, ensuring data is sorted, deduplicated, and formatted correctly.
Annotate features on the identified plasmids using mobile genetic element database annotations.
Construct a final annotated dataset integrating genetic element information for downstream applications.
Time estimation: 1 hourLevel: Introductory IntroductorySupporting Materials:Published: Jan 7, 2025Last modification: Apr 13, 2026License: Tutorial Content is licensed under Creative Commons Attribution 4.0 International License. The GTN Framework is licensed under MITpurl PURL: https://gxy.io/GTN:T00501version Revision: 2
Identifying and annotating mobile genetic elements (MGE) in metagenomic data can be tricky. To facilitate the process, we can use existing and custom annotated mobile genetic element database.
The metaplasmidome refers to the collection of all plasmids present within a given environment, typically identified through metagenomic sequencing. Plasmids are extrachromosomal genetic elements that often carry genes associated with antibiotic resistance, virulence, or metabolic functions, making them crucial for microbial adaptability. In the context of metagenomics, plasmids are identified alongside chromosomal DNA.
A common step in metaplasmidome analysis is searching sequencing reads against known plasmid databases to detect plasmid sequences within a metagenome, allowing researchers to map the diversity and abundance of plasmids in various environments.
In this tutorial, we use a metaplasmidome database built from public air metagenomes and query it with assembled air metagenome data. A similar approach can be used for other mobile genetic elements.
The air metaplasmidome is available from Zenodo (DEBROAS 2024) and was built from metagenomic data selected in Web of Science (Clarivate) on October 2022 using keywords:
txid655179[Organism:noexp] AND metagenome [Filter]; AIR Metagenome; Air microbiome; Troposphere; Aerosol; Atmosphere. Data were manually curated to remove sequencing originated from metabarcoding data (i.e., 16S). The assembled data supplied by MetaSUB consortium (Danko et al. 2021) when available was used for air metagenome in the built environments.Plasmid contents were predicted using the assembled data. Metagenomes sequencing by Illumina (paired-illumina reads) were assembled by using MEGAHIT 1.2.9 with metalarge option (Li et al. 2015) after cleaning data with bbduk2 (qtrim=rl trimq=28 minlen=25 maq=20 ktrim=r k=25 mink=11 and a list of adaptators to remove) from bbtools suite
Plasmids were predicted for each assembling by using PlasSuite scripts describing in-depth in Hilpert et al. Hilpert et al. 2021 Hennequin et al. 2022 and available on GitHub. Briefly, contigs were analyzed using both reference-based and reference-free approaches. The databases employed included those for chromosomes (archaea and bacteria) and plasmids from NCBI, as well as the MOB-suite tool (Robertson and Nash 2018), SILVA (Quast et al. 2012) and phylogenetic markers harbored by chromosomes (Wu et al. 2013). Two reference-free methods were applied to contigs that were not affiliated with chromosomes (discarded) or plasmids (retained in the first step): PlasFlow (Krawczyk et al. 2018) and PlasClass (Pellow et al. 2020). Viruses were removed by using viralVerify (Antipov et al. 2020) that provides in parallel provide plasmid/non-plasmid classification.
Eukaryotes contaminants were removed by aligning the sequences against NT databases and human chromosomes (GRCh38) with minimap2 with -x asm5 option (Li 2018). Contigs mapping with an identity of 95% and a coverage of 80% were removed. the final plasmidome set was clustered by mmseqs (Mirdita et al. 2019) with 80% of coverage and 90% of identity (–min-seq-id 0.90 -c 0.8 –cov-mode 1 –cluster-mode 2 –alignment-mode 3 –kmer-per-seq-scale 0.2).
AgendaIn this tutorial, we will cover:
Galaxy and data preparation
Any analysis should get its own Galaxy history. So let’s start by creating a new history and import the data (plasmidome database and query metagenomes) into it.
Hands On: Prepare Galaxy and data
Create a new history for this tutorial
To create a new history simply click the new-history icon at the top of the history panel:
Rename the history
- Click on galaxy-pencil (Edit) next to the history name (which by default is “Unnamed history”)
- Type the new name
- Click on Save
- To cancel renaming, click the galaxy-undo “Cancel” button
If you do not have the galaxy-pencil (Edit) next to the history name (which can be the case if you are using an older version of Galaxy) do the following:
- Click on Unnamed history (or the current name of the history) (Click to rename history) at the top of your history panel
- Type the new name
- Press Enter
Import the metaplasmidome reference database from Zenodo or from the shared data library
https://zenodo.org/records/14501567/files/air_metaplasmidome.fasta.gz
- Copy the link location
Click galaxy-upload Upload at the top of the activity panel
- Select galaxy-wf-edit Paste/Fetch Data
Paste the link(s) into the text field
Press Start
- Close the window
As an alternative to uploading the data from a URL or your computer, the files may also have been made available from a shared data library:
- Go into Libraries (left panel)
- Navigate to the correct folder as indicated by your instructor.
- On most Galaxies tutorial data will be provided in a folder named GTN - Material –> Topic Name -> Tutorial Name.
- Select the desired files
- Click on Add to History galaxy-dropdown near the top and select as Datasets from the dropdown menu
In the pop-up window, choose
- “Select history”: the history you want to import the data to (or create a new one)
- Click on Import
Rename
Air plasmidome database
- Click on the galaxy-pencil pencil icon for the dataset to edit its attributes
- In the central panel, change the Name field
- Click the Save button
Import the reads to query against the reference database from Zenodo or from the shared data library
https://zenodo.org/records/14501567/files/air_metagenome_assemblies.fasta.gzRename
Air metagenomes


Question
- How many reads are in the query dataset?
- How many plasmid sequences are in the metaplasmidome database?
- 16,637 sequences
- 674,495 sequences
Read mapping against the metaplasmidome database
Hands On: Map reads against metaplasmidome database
- Map with minimap2 ( Galaxy version 2.28+galaxy0) with the following parameters:
- “Will you select a reference genome from your history or use a built-in index?”:
Use a genome from history and build index
- param-file “Use the following dataset as the reference sequence”:
metaplasmidome database- “Single or Paired-end reads”:
Single
- param-file “Select fastq dataset”:
query- “Select a profile of preset options”:
Long assembly to reference mapping (-k19 -w19 -A1 -B19 -O39,81 -E3,1 -s200 -z200 --min-occ-floor=100). Typically, the alignment will not extend to regions with 5% or higher sequence divergence. Only use this preset if the average divergence is far below 5%. (asm5)- In “Set advanced output options”:
- “Select an output format”:
PAF
PAF is the default output format of minimap2. It is TAB-delimited with each line consisting of the following predefined fields:
- Query sequence name
- Query sequence length
- Query start coordinate (0-based)
- Query end coordinate (0-based)
+if query/target on the same strand and-if opposite- Target sequence name
- Target sequence length
- Target start coordinate on the original strand
- Target end coordinate on the original strand
- Number of matching bases in the mapping
- Number bases, including gaps, in the mapping
- Mapping quality
Question
- How many lines are in the file?
- Can a query sequence be found several times in the target?
- Are all alignments of good quality?
- 29,796 lines
- There are 16,637 query sequences. Several query sequences are found several times in the file: SRR17300492_75807 is found 16 times. It means they have been mapped to several locations or sequences in the target.
- The first alignment (SRR17300492_75807 mapping on SRR17300667-707) has a score of 60, the highest Phred score. The third alignment has a score of 0, so not good.
Exploration of the mapping results
Let’s look at the distribution of the mapping score (column 12)
Hands On: Plot score distribution
Change datatype to tabular
- Click on the galaxy-pencil pencil icon for the dataset to edit its attributes
- In the central panel, click galaxy-chart-select-data Datatypes tab on the top
- In the galaxy-chart-select-data Assign Datatype, select your desired datatype from “New Type” dropdown
- Tip: you can start typing the datatype into the field to filter the dropdown menu
- Click the Save button
- Cut columns from a table with:
- “Cut columns”:
c12- “Delimited by”:
tab- param-file “From”: Output of Map with minimap2 tool
- Histogram with ggplot2 ( Galaxy version 3.4.0+galaxy0) with the following parameters:
- “Input should have column headers - these will be the columns that are plotted * “: Output of Cut tool
- “Label for x axis”:
Score- “Label for y axis”:
Distribution- In “Advanced Options”:
- “Legend options”:
Hide legend
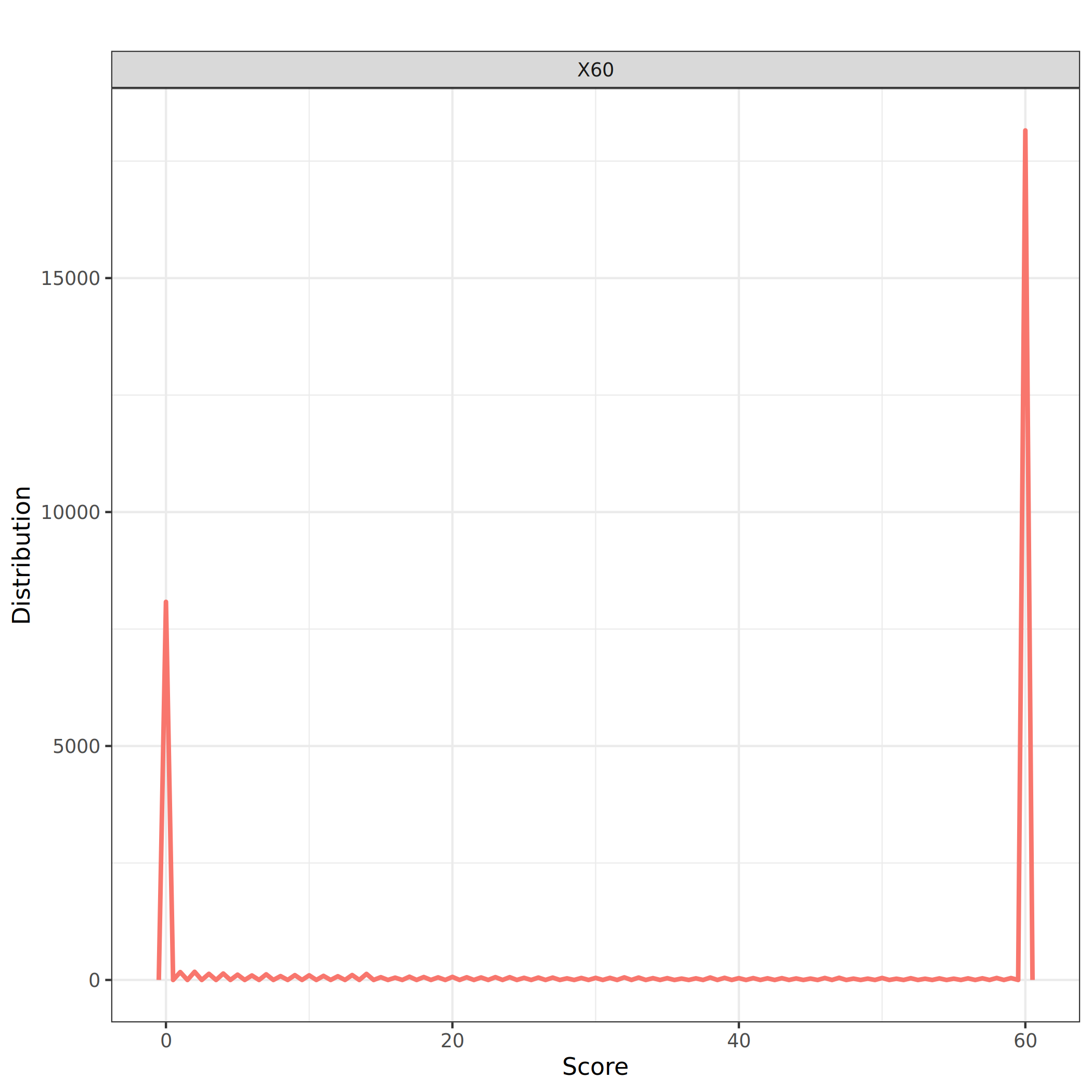
QuestionAre all alignments of good quality?
There is a pic at 60 but many alignments have a score below 40.
We should remove alignments with a score below 40. Before that, let’s look at the plasmid coverage. We first need to compute it, i.e. the ratio between the number of matching bases in the mapping (Column 10) and plasmid length (target sequence length - Column 7).
Hands On: Compute plasmid coverage
- Cut columns from a table with:
- “Cut columns”:
c1-c12- “Delimited by”:
tab- param-file “From”: Output of Map with minimap2 tool
- Compute on rows ( Galaxy version 2.1) with the following parameters:
- “Input file”: Output of Cut tool
- In “Expressions”:
- “Add expression”:
float(c10)/float(c7)- Rename
Mapping stats + plasmid coverage
A new column has been added (column 13) with the plasmid coverage. Let’s now plot its distribution.
Hands On: Plot plasmid coverage distribution
- Cut columns from a table with:
- “Cut columns”:
c13- “Delimited by”:
tab- param-file “From”:
Mapping stats + plasmid coverage- Histogram with ggplot2 ( Galaxy version 3.4.0+galaxy0) with the following parameters:
- “Input should have column headers - these will be the columns that are plotted * “: Output of Cut tool
- “Label for x axis”:
Plasmid coverage- “Label for y axis”:
Distribution- “Bin width for plotting”:
0.1- In “Advanced Options”:
- “Legend options”:
Hide legend
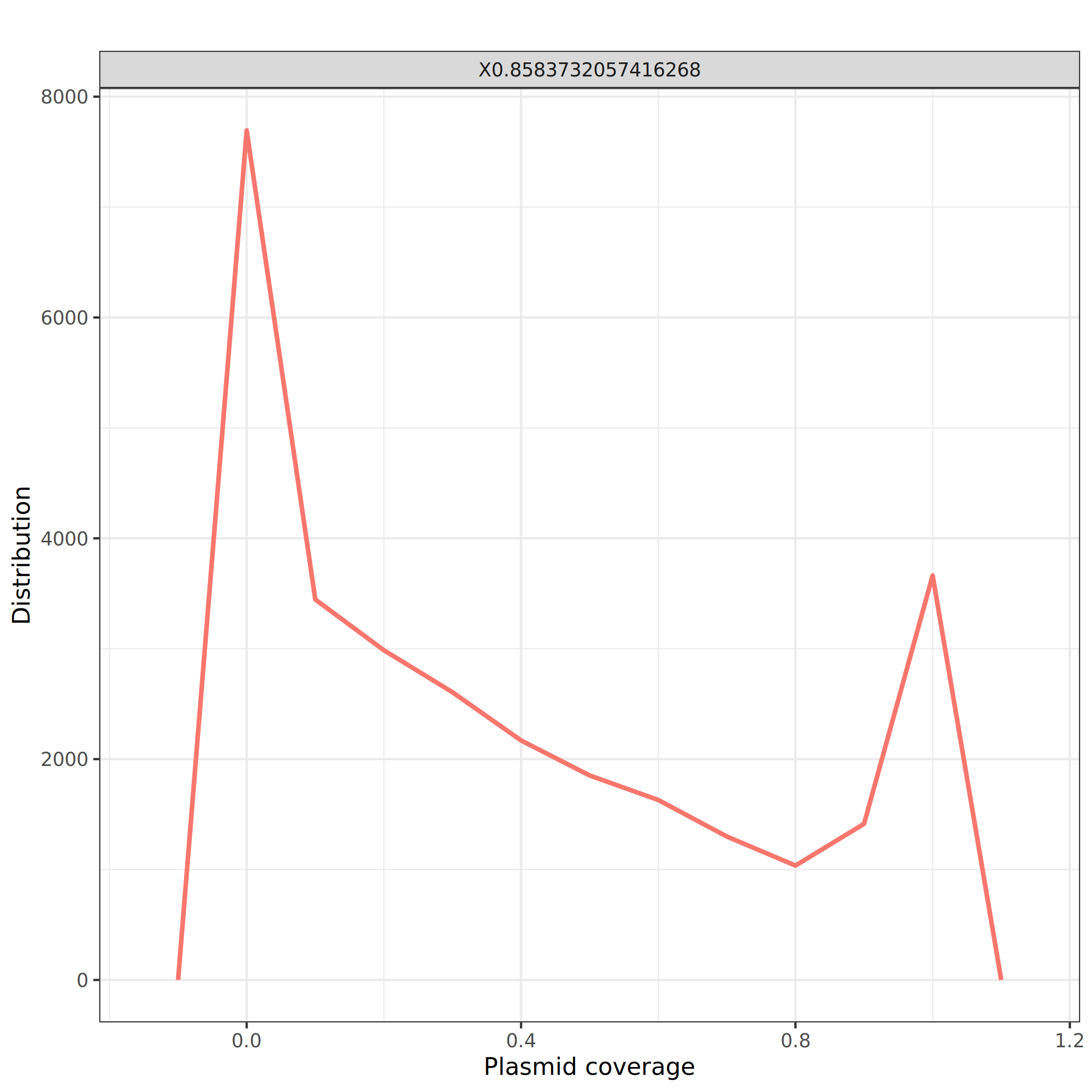
Question
- What is the distribution of the plasmid coverage?
- What could be a good threshold to filter?
- There are 2 peaks: one around 0 (i.e. no plasmid coverage) that slowly decreases until 0.8 and a pic at 1 (full plasmid coverage)
- 0.8 seems to be a breaking point and could be a good value to filter.
Filtering
We will now filter the alignment to keep only the ones with a plasmid coverage (column 13) of at least 0.8, i.e. a read mapping to a plasmid covering at least 80% of the plasmid.
Hands On: Filter alignments based on plasmid coverage
- Filter data on any column using simple expressions with the following parameters:
- param-file “Filter”:
Mapping stats + plasmid coverage- “With following condition”:
c13>=0.8- Rename
Alignments with plasmid coverage >= 0.8
Question
- How many lines have been kept?
- Which percentage of lines does that correspond to?
- What does the distribution of the mapping score look like for these alignments?
- Is there any extra filter we should do on the data?
- 5,577 (over the 29,796)
- 18.73%
To plot the distribution of the mapping score for filtered alignments, we need to run the series of tools as done earlier:
- Cut columns from a table with:
- “Cut columns”:
c12- “Delimited by”:
tab- param-file “From”:
Alignments with plasmid coverage >= 0.8- Histogram with ggplot2 ( Galaxy version 3.4.0+galaxy0) with the following parameters:
- “Input should have column headers - these will be the columns that are plotted * “: Output of Cut tool
- “Label for x axis”:
Score- “Label for y axis”:
Distribution- In “Advanced Options”:
- “Legend options”:
Hide legend
Most of the alignments are of good quality (pic at 60). There is also a pic at 0, meaning that some alignments are of bad quality
- It might be good to add a filter on the score.
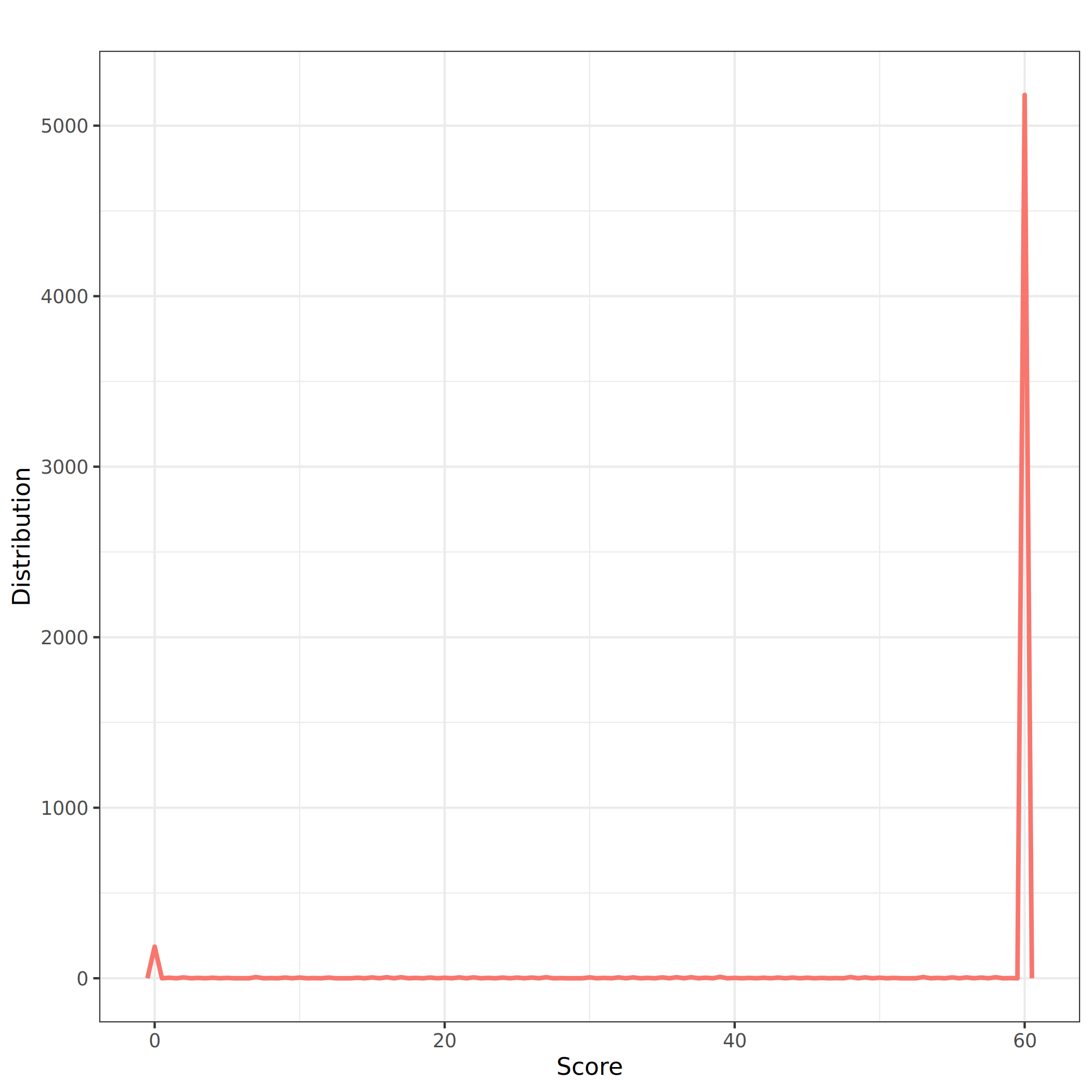
After filtering on the plasmid coverage, we also add a filter on the score to be sure that we keep only the best alignments (i.e. a score - column 12 - of at least 40).
Hands On: Filter alignments based on score
- Filter data on any column using simple expressions with the following parameters:
- param-file “Filter”:
Alignments with plasmid coverage >= 0.8- “With following condition”:
c12>=40- Rename
Alignments with plasmid coverage >= 0.8 and score >= 40
Question
- How many lines have been kept?
- Which percentage of lines does that correspond to?
- 5,249 (over the 5,577)
- 94.12%
Extract sequences mapping to plasmids
Let’s now extract the sequences mapping to plasmids (coverage higher than 80%).
First, we need to extract the names of the reads (in air metagenomes) mapping to plasmids in the metaplasmidome database. For that, we need to extract column 1 (query names, i.e. names of reads in Air metagenomes) and 6 (reference names, i.e. names of sequences in Air plasmidome database).
Hands On: Get sequences matching to plasmids
- Cut with the following parameters:
- “Cut columns”:
c1,c6- param-file “From”:
Alignments with plasmid coverage >= 0.8 and score >= 40Rename to
Names for alignments with plasmid coverage >= 0.8 and score >= 40- Sort with the following parameters:
- param-file “Sort Dataset”: output of Cut tool
- “on column”:
c2
Second, we need to convert the Air metagenomes FASTA file to a tabular so we can join it with Names for alignments with plasmid coverage >= 0.8 and score >= 40
Hands On: Get query sequence as a table
- Convert FASTA to Tabular with the following parameters:
- param-file “Fasta file”:
Air metagenomes- Rename to
Air metagenome as table
Let’s now join the 2 datasets on the names of the reads in air metagenomes so column 1 in Names for alignments with plasmid coverage >= 0.8 and score >= 40 and column 1 in Air metagenome as table.
Hands On: Get sequences matching to plasmids
- Join two Datasets with the following parameters:
- param-file “Join”: output of Sort tool
- “using column”:
Column: 1- param-file “with”: output of Convert FASTA to Tabular tool
- “and column”:
Column: 1- “Fill empty columns”:
No
We have now a table with 4 columns: names of the reads in air metagenomes, names of sequences in the reference database, names of the reads in air metagenomes, sequence of reads in air metagenomes. We will now create 2 outputs:
- A table with metaplasmidome database sequence name, metagenomic sequence names and the metagenomic sequences
- A FASTA file with metagenomic sequences
Create the table with air metagenomes identified as plasmids
Let’s remove the duplicated column (column 3) and reorganize the columns
Hands On: Keep non duplicated columns
- Cut with the following parameters:
- “Cut columns”:
c2,c1,c4- param-file “From”: output of Join two Datasets tool
Let’s now add column names to the generated table.
Hands On: Add column names
- Add Header ( Galaxy version 0.1.3) with the following parameters:
- “List of Column headers”:
Metaplasmidome database sequence name,Metagenomic sequence name,Metagenomic sequence- param-file “Data File (tab-delimted)”: output of Replace Text tool
- Rename the output to
Air metagenomes identified as plasmids
Create the FASTA file with air metagenomes identified as plasmids
We can now generate a FASTA file with the identified sequences.
From the output of Join two Datasets tool, let’s keep only metagenomic sequence names and the metagenomic sequences and remove duplicated sequences
Hands On: Keep unique metagenomic sequences
- Cut with the following parameters:
- “Cut columns”:
c1,c4- param-file “From”: output of Join two Datasets tool
- Sort with the following parameters:
- param-file “Sort Dataset”: output of Cut tool
- “on column”:
Column: 1- “with flavor”:
Alphabetical sort- Unique occurrences of each record ( Galaxy version 9.3+galaxy1) with the following parameters:
- param-file “File to scan for unique values”: output of Sort tool
QuestionHow many lines have been kept?
4,055 (over 5,249)
We have now a table with unique metagenomic sequences. Let’s transform it into a FASTA file.
Hands On: Convert to a FASTA file
- Tabular-to-FASTA ( Galaxy version 1.1.1) with the following parameters:
- param-file “Tab-delimited file”: output of Unique occurrences of each record tool
- “Title column(s)”:
Column: 1- “Sequence column”:
Column: 2- Rename the output to
Air metagenome sequences identified as plasmids
Annotate features on the identified plasmids
Let’s now annotate features on the identified plasmids. For that, we will use the annotation of the air metaplasmidome sequences that have been done with Prokka (citation) and assume that the annotations are similar for the identified plasmids.
We will import the GFF generated by Prokka.
Hands On: Import the GFF with metaplasmidome reference database annotation
Import the metaplasmidome reference database annotation from Zenodo or from the shared data library
https://zenodo.org/records/14501567/files/air_metaplasmidome_annotations.gffInspect it.
This file is a GFF: it describes genes and other features of DNA, RNA and protein sequences. It is a tab-delimited file with 9 fields per line:
- seqid: The name of the sequence where the feature is located.
- source: The algorithm or procedure that generated the feature.
- type: The feature type name, like “gene” or “exon”.
- start: Genomic start of the feature, with a 1-base offset.
- end: Genomic end of the feature, with a 1-base offset.
- score: Numeric value that generally indicates the confidence of the source in the annotated feature.
- strand: Single character that indicates the strand of the feature.
- phase: Phase of CDS features.
- attributes: A list of tag-value pairs separated by a semicolon with additional information about the feature.
The seqid corresponds here to the ID of the sequences in the metaplasmidome reference database. So to filter, we need to compare Metaplasmidome database sequence name in Air metagenomes identified as plasmids
to seqid by joining the 2 datasets on column 1.
The file has with ~85 Million lines but many are comments (lines starting with ##).
QuestionHow many features are in the GFF file?
2,951,015 features
To get this number, we run Count GFF Features ( Galaxy version 0.2) with the following parameters:
- param-file “GFF Dataset to Filter”: imported GFF
Let’s now filter the GFF to keep only information related to the sequences matching to plasmids.
For that, we join the GFF on the SeqID column (column 1) with the Air metagenomes identified as plasmids file on the ` Metaplasmidome database sequence name` (Column 1)
Hands On: Extract information about the sequences matching to plasmids
- Join two Datasets with the following parameters:
- param-file “Join”: imported GFF
- “using column”:
Column: 1- param-file “with”:
Air metagenomes identified as plasmids- “and column”:
Column: 1- “Fill empty columns”:
No- Inspect the generated file
Question
- How many lines have been kept?
- Why is there more lines than in the
Air metagenomes identified as plasmidsfile?- What are the columns?
- Which columns should we keep if we want to keep the Metagenomic sequence name, the feature and the attributes?
- 26k+ lines
- Some sequences (e.g. SRR17300493-2380) have several lines: several features (CDS, rRNA, etc) annotated on it
- The 9 columns of the GFF file + the 3 columns of the
Air metagenomes identified as plasmidsfile- Columns to keep:
- Metagenomic sequence name (Column 11)
- Feature (Column 3)
- Attributes (Column 9)
Let’s now cut the columns.
Hands On: Cut and filter
- Cut with the following parameters:
- “Cut columns”:
c11,c3,c9- param-file “From”: output of Join two Datasets tool
- Inspect the generated file
QuestionWhat are the different identified features?
Using Group data by a column to group and count on 2nd column, we find 26,329 CDS and 307 tRNA.
Extract the CDS
Let’s filter to keep only the CDS and extract the gene names.
Hands On: Keep CDS
- Filter with the following parameters:
- param-file “Filter”: output of Cut tool
- “With following condition”:
c2=='CDS'- Inspect the generated file
We have now a file with 26,329 lines. The 2nd column is not useful so we will remove it. Column 3 (atributes in GFF) lists tag-value pairs separated by a semicolon with additional information about the feature.
The first lines seems to be hypothetical proteins. If we scroll down, we can find some annotated genes (with gene keyword).
It would be good to create a tabular file with:
- Metagenomic sequence name
- Gene ID
- Gene name
- Gene product
As not all genes are annotated (with gene keyword), we first need to split the file between hypothetical proteins and annotated genes and process the two generated files independently.
Hands On: Keep CDS
- Cut with the following parameters:
- “Cut columns”:
c1,c3- param-file “From”: output of Filter tool
- Filter by keywords and/or numerical value ( Galaxy version 2021.04.19.1) with the following parameters:
- param-file “Input file”: output of last Cut tool
- “Does file contain header? “:
No- In Filter by keywords:
- “Column number on which to apply the filter”:
c2- In
Enter keywords:
copy/paste- “Copy/paste keywords to find (keep or discard)”:
gene=- Inspect the generated files
Question
- How many lines have been kept (non-hypothetical proteins) and how many have been discarded (hypothetical proteins)?
- Which information do we have for the 2nd identified gene?
- There are:
- 25,001 hypothetical proteins CDS (lines in the discarded lines file)
- 1,329 annotated CDS (lines in the other file)
- For the 2nd gene, the column 3 is
ID=BAFOEJEB_01977;eC_number=1.8.5.7;Name=yqjG_1;dbxref=COG:COG0435;gene=yqjG_1;inference=ab initio prediction:Prodigal:2.6,similar to AA sequence:UniProtKB:P42620;locus_tag=BAFOEJEB_01977;product=Glutathionyl-hydroquinone reductase YqjG, which mean:
- Gene name is
yqjG_1- The product is
Glutathionyl-hydroquinone reductase YqjG
Let’s extract gene ID, gene name and the product in different columns from the annotated gene output and add a header to the file.
Hands On: Prepare the annotated CDS file
Change annotated CDS output of Filter by keywords tool datatype to tabular
- Click on the galaxy-pencil pencil icon for the dataset to edit its attributes
- In the central panel, click galaxy-chart-select-data Datatypes tab on the top
- In the galaxy-chart-select-data Assign Datatype, select your desired datatype from “New Type” dropdown
- Tip: you can start typing the datatype into the field to filter the dropdown menu
- Click the Save button
- Replace Text in a specific column ( Galaxy version 9.3+galaxy1) with the following parameters:
- param-file “File to process”: output of Filter by keywords tool
- In Replacement:
- “in column”:
Column: 2- “Find pattern”:
ID=([^;]*);.*;gene=([^;]*).*;product=([^;]*).*- “Replace with”:
\\1\t\\2\t\\3- Add Header ( Galaxy version 0.1.3) with the following parameters:
- “List of Column headers”:
Metagenomic sequence name,Gene ID,Gene name,Gene product- param-file “Data File (tab-delimited)”: output of Replace Text tool
Let’s now prepare the hypothetical protein CDS file
Hands On: Prepare the hypothetical CDS gene file
Change hypothetical CDS gene output of Filter by keywords tool datatype to tabular
Replace Text in a specific column ( Galaxy version 9.3+galaxy1) with the following parameters:
- param-file “File to process”: output of Filter by keywords tool
- In Replacement:
- “in column”:
Column: 2- “Find pattern”:
ID=([^;]*);.*- “Replace with”:
\\1\t\t
We can now merge both files.
Hands On: Concatenate the annotated and hypothetical protein gene files
- Concatenate multiple datasets or collections with the following parameters:
- param-file “Concatenate Dataset”: output of Add Header tool
- In Dataset:
- param-file “Select”: output of last Replace Text tool
- Rename
CDS in metagenomes identified as plasmids
Add KO and PFAM annotation
Let’s expand annotation with KO (KEGG Orthology) (Kanehisa et al. 2016), a database of molecular functions represented in terms of functional orthologs, and PFAM (Mistry et al. 2021), a large collection of protein families. The files are
Hands On: Import KO and PFAM annotations
Import the KO and PFAM annotations from Zenodo or from the shared data library
https://zenodo.org/records/14501567/files/KOsignificatifNR.tsv https://zenodo.org/records/14501567/files/pfamsignificatifNR.tsvInspect both files
Both are tabular files with several columns including the gene ID, KO/PFAM ID, and some extra annotation.
Let’s now join the files with Annotated genes in air metagenomes sequences identified as plasmids to extend the annotations using gene ID.
Hands On: Join with KO and PFAM annotation files
- Join two Datasets with the following parameters:
- param-file “Join”:
Annotated genes in air metagenomes sequences identified as plasmids- “using column”:
Column: 2- param-file “with”: imported KO file
- “and column”:
Column: 2- “Keep lines of first input that do not join with second input”:
YesInspect the generated file
- Cut with the following parameters:
- “Cut columns”:
c1-c4,c7,c11- param-file “From”: output of Join tool
- Join two Datasets with the following parameters:
- param-file “Join”: output of Cut tool
- “using column”:
Column: 2- param-file “with”: imported PFAM file
- “and column”:
Column: 1- “Keep lines of first input that do not join with second input”:
YesInspect the generated file
- Cut with the following parameters:
- “Cut columns”:
c1-c6,c9,c10,c25- param-file “From”: output of Join tool
- Select last lines from a dataset (tail) ( Galaxy version 9.3+galaxy1) with the following parameters:
- param-file “Text file”: output of Cut tool
- “Operation”:
Keep everything from lines on- “Number of lines”:
2- Add Header ( Galaxy version 0.1.3) with the following parameters:
- “List of Column headers”:
Metagenomic sequence name,Gene ID,Gene name,Gene product,KO ID,KO annotation,PFAM name,PFAM ID,PFAM annotation- param-file “Data File (tab-delimited)”: output of Cut tool
- Rename
CDS in metagenomes identified as plasmids + KO + PFAM
Let’s look at the KO and PFAM statistics.
Question
- How many genes have been extended with KO information?
- How many different KO are found?
- Which KO is the most found?
- How many genes have been extended with PFAM information?
- How many different PFAM are found?
- Which PFAM is the most found?
- Using Filter with
c5!='.', we find that 426 CDS (1.62%) have been extended with KO information.- 277 KO ( Group data by a column to group and count on 5th column)
- K14572 (Ribosome biogenesis in eukaryotes) is found 9 times ( Sort by descending order on the Group data by a column output on column 2)
- 4,817 CDS (18.29%) have been extended with PFAM information ( Filter with
c8!='.'onCDS + KO + PFAM)- 410 PFAM ( Group data by a column to group and count on 8th column)
- PF00961.22 (Cytochrome c oxidase subunit 1) is found 89 times ( Sort by descending order)
Let’s extract an overview of the annotations per metagenomic sequence by grouping on the 1st column and counting the number of distinct values on gene ID, gene name, KO ID, PFAM ID.
Hands On: Annotation per metagenomic sequences
- Group data by a column with the following parameters:
- param-file “Select data”:
CDS + KO + PFAM- “Group by column”:
Column: 1- In “Operation”:
- In “1: Operation”:
- “Type”:
Count distinct- “On column”:
Column: 2- In “2: Operation”:
- “Type”:
Count distinct- “On column”:
Column: 3- In “3: Operation”:
- “Type”:
Count distinct- “On column”:
Column: 5- In “4: Operation”:
- “Type”:
Count distinct- “On column”:
Column: 8- Select last lines from a dataset (tail) ( Galaxy version 9.3+galaxy1) with the following parameters:
- param-file “Text file”: output of Cut tool
- “Operation”:
Keep everything from lines on- “Number of lines”:
2- Add Header ( Galaxy version 0.1.3) with the following parameters:
- “List of Column headers”:
Metagenomic sequence name,Number of CDS,Number of annotated CDS,Number of associated KO,Number of associated PFAM- param-file “Data File (tab-delimited)”: output of Select last tool
- Rename
CDS annotation overview per metagenomic sequences
Conclusion
You've Finished the Tutorial
Key points
Leveraging annotated mobile genetic element databases, such as the metaplasmidome, is essential for identifying and characterizing plasmids and other genetic elements in metagenomic data.
Effective filtering based on plasmid coverage and mapping quality is critical to ensure the reliability and accuracy of extracted sequences.
Generating well-organized outputs, such as FASTA files and annotated tables, is vital for downstream analysis and collaborative research.
Annotating genetic features on identified plasmids enhances understanding of their biological roles, such as antibiotic resistance or virulence.
Frequently Asked Questions
Have questions about this tutorial? Have a look at the available FAQ pages and support channelsUseful literature
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
References
- Quast, C., E. Pruesse, P. Yilmaz, J. Gerken, T. Schweer et al., 2012 The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic acids research 41: D590–D596. 10.1093/nar/gks1219
- Wu, D., G. Jospin, and J. A. Eisen, 2013 Systematic identification of gene families for use as “markers” for phylogenetic and phylogeny-driven ecological studies of bacteria and archaea and their major subgroups. PloS one 8: e77033. 10.1371/journal.pone.0077033
- Li, D., C.-M. Liu, R. Luo, K. Sadakane, and T.-W. Lam, 2015 MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31: 1674–1676. 10.1093/bioinformatics/btv033
- Kanehisa, M., Y. Sato, M. Kawashima, M. Furumichi, and M. Tanabe, 2016 KEGG as a reference resource for gene and protein annotation. Nucleic acids research 44: D457–D462. 10.1093/nar/gkv1070
- Krawczyk, P. S., L. Lipinski, and A. Dziembowski, 2018 PlasFlow: predicting plasmid sequences in metagenomic data using genome signatures. Nucleic acids research 46: e35–e35. 10.1093/nar/gkx1321
- Li, H., 2018 Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191
- Robertson, J., and J. H. E. Nash, 2018 MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microbial genomics 4: e000206. 10.1099/mgen.0.000206
- Mirdita, M., M. Steinegger, and J. Söding, 2019 MMseqs2 desktop and local web server app for fast, interactive sequence searches. Bioinformatics 35: 2856–2858. 10.1093/bioinformatics/bty1057
- Pellow, D., I. Mizrahi, and R. Shamir, 2020 PlasClass improves plasmid sequence classification. PLoS computational biology 16: e1007781. 10.1371/journal.pcbi.1007781
- Danko, D., D. Bezdan, E. E. Afshin, S. Ahsanuddin, C. Bhattacharya et al., 2021 A global metagenomic map of urban microbiomes and antimicrobial resistance. Cell 184: 3376–3393. 10.1016/j.cell.2021.05.002
- Hilpert, C., G. Bricheux, and D. Debroas, 2021 Reconstruction of plasmids by shotgun sequencing from environmental DNA: which bioinformatic workflow? Briefings in bioinformatics 22: bbaa059. 10.1093/bib/bbaa059
- Mistry, J., S. Chuguransky, L. Williams, M. Qureshi, G. A. Salazar et al., 2021 Pfam: The protein families database in 2021. Nucleic acids research 49: D412–D419. 10.1093/nar/gkaa913
- Hennequin, C., C. Forestier, O. Traore, D. Debroas, and G. Bricheux, 2022 Plasmidome analysis of a hospital effluent biofilm: Status of antibiotic resistance. Plasmid 122: 102638. 10.1016/j.plasmid.2022.102638
- DEBROAS, D., 2024 Plasmids Identified in Air Metagenomes. 10.5281/zenodo.11124657
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.

Citing this Tutorial
- Bérénice Batut, Nadia Goué, Didier Debroas, Query an annotated mobile genetic element database to identify and annotate genetic elements (e.g. plasmids) in metagenomics data (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/microbiome/tutorials/metaplasmidome_query/tutorial.html Online; accessed TODAY
- Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
Congratulations on successfully completing this tutorial!@misc{microbiome-metaplasmidome_query, author = "Bérénice Batut and Nadia Goué and Didier Debroas", title = "Query an annotated mobile genetic element database to identify and annotate genetic elements (e.g. plasmids) in metagenomics data (Galaxy Training Materials)", year = "", month = "", day = "", url = "\url{https://training.galaxyproject.org/training-material/topics/microbiome/tutorials/metaplasmidome_query/tutorial.html}", note = "[Online; accessed TODAY]" } @article{Hiltemann_2023, doi = {10.1371/journal.pcbi.1010752}, url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752}, year = 2023, month = {jan}, publisher = {Public Library of Science ({PLoS})}, volume = {19}, number = {1}, pages = {e1010752}, author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and}, editor = {Francis Ouellette}, title = {Galaxy Training: A powerful framework for teaching!}, journal = {PLoS Comput Biol} }
You can use Ephemeris's
shed-tools installcommand to install the tools used in this tutorial.shed-tools install [-g GALAXY] [-a API_KEY] -t <(curl https://training.galaxyproject.org/training-material/api/topics/microbiome/tutorials/metaplasmidome_query/tutorial.json | jq .admin_install_yaml -r)Alternatively you can copy and paste the following YAML
--- install_tool_dependencies: true install_repository_dependencies: true install_resolver_dependencies: true tools: - name: text_processing owner: bgruening revisions: 86755160afbf tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: text_processing owner: bgruening revisions: 86755160afbf tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: text_processing owner: bgruening revisions: 86755160afbf tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: column_maker owner: devteam revisions: aff5135563c6 tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: count_gff_features owner: devteam revisions: 188392a0d0a8 tool_panel_section_label: Statistics tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: histogram owner: devteam revisions: 6f134426c2b0 tool_panel_section_label: Graph/Display Data tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: tabular_to_fasta owner: devteam revisions: 0a7799698fe5 tool_panel_section_label: FASTA/FASTQ tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: add_column_headers owner: estrain revisions: ff2acdb98a74 tool_panel_section_label: Microbiome tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: ggplot2_histogram owner: iuc revisions: b0d96516e6a5 tool_panel_section_label: Graph/Display Data tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: minimap2 owner: iuc revisions: 5cc34c3f440d tool_panel_section_label: Mapping tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: proteore_filter_keywords_values owner: proteore revisions: 98cb671a92eb tool_panel_section_label: Microbiome tool_shed_url: https://toolshed.g2.bx.psu.edu/