The development of medicinal chemistry is advancing very rapidly. Big pharmaceutical companies, research institutes and universities are working on ground-breaking solutions to help patients combat all kinds of diseases. During that development process, tons of data are generated – not only from the lab environment but also from clinical trials. Given that the discovery of more potent, safer and cheaper drugs is the ultimate goal of all research bodies, we should all focus on making the data we gather FAIR: Findable, Accessible, Interoperable, and Reusable to push the boundaries of drug development even further.
With the currently available methods such as artificial intelligence, machine learning, many toolkits, software and access to various databases, managing big data is now inherently linked to medicinal chemistry and helps to make this area as efficient as it can be.
In this tutorial, we will therefore explore some concepts related to medicinal chemistry, explore the available chemical and pharmacological databases, and perform some basic data analyses using the Galaxy interface.
There are many factors that medicinal chemists take into account while designing new drugs. It is essential to estimate the properties of the molecule before synthesising it in the lab, so the structures of drug candidates are usually compared to the existing drugs by so-called drug-likeness. This includes intrinsic properties of a compound that will lead to favourable ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) characteristics. Below are some properties usually assessed:
Lipophilicity
Size
Polarity
Insolubility
Unsaturation
Flexibility
Additionally, there have been some rules developed to help estimate drug-likeness, amongst which:
As you see, there are a number of characteristics to consider before even synthesising the molecule in a lab. One of the online tools that help to summarise all the most important information about intrinsic properties and drug-likeness of compounds is called SwissADME and is a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules (Daina et al. 2017).
Below we briefly discuss two probably the most well-known rules: Lipinski’s and Veber’s rules.
The Lipinski rule of 5
This rule of thumb developed back in 1997 by Christopher Lipinski and colleagues tries to predict the likelihood that a given small molecule can be orally active. The Lipinski rule of 5 favours molecules as potential oral drug candidates if:
the molecular mass is less than 500
the calculated logarithm of the octanol−water partition coefficient (clogP) is less than 5
they have up to 5 H-bond donors
they have up to 10 H-bond acceptors
However, nowadays there are more and more drugs being developed which don’t comply with those rules and regardless are still effective. There are voices from the scientific community, pointing out that “We are in danger of repeating our past mistakes if we assume these new modalities are not ‘drug-like’ and cannot be oral drugs because they are not [rule of 5] compliant” (Michael Shultz from Novartis, cited in Mullard 2018 where the authors re-assess the rule of 5). Then, in O′Hagan et al. 2014 we read “This famous “rule of 5” has been highly influential in this regard, but only about 50 % of orally administered new chemical entities actually obey it.”
Veber’s rule
In (Veber et al. 2002) we read that the commonly applied molecular weight cutoff at 500 does not itself significantly separate compounds with poor oral bioavailability from those with acceptable values. The authors explain that on average both the number of rotatable bonds and polar surface area or hydrogen bond count tend to increase with molecular weight which may in part explain the success of the molecular weight parameter in predicting oral bioavailability. Their observations suggest that compounds will have a high probability of good oral bioavailability if they meet the two criteria:
10 or fewer rotatable bonds - increased rotatable bond count has a negative effect on the permeation rate
a polar surface area no greater than 140 Å 2 (or 12 or fewer H-bond donors and acceptors) - reduced polar surface area correlates better with increased permeation rate than does lipophilicity
Big data for drug discovery
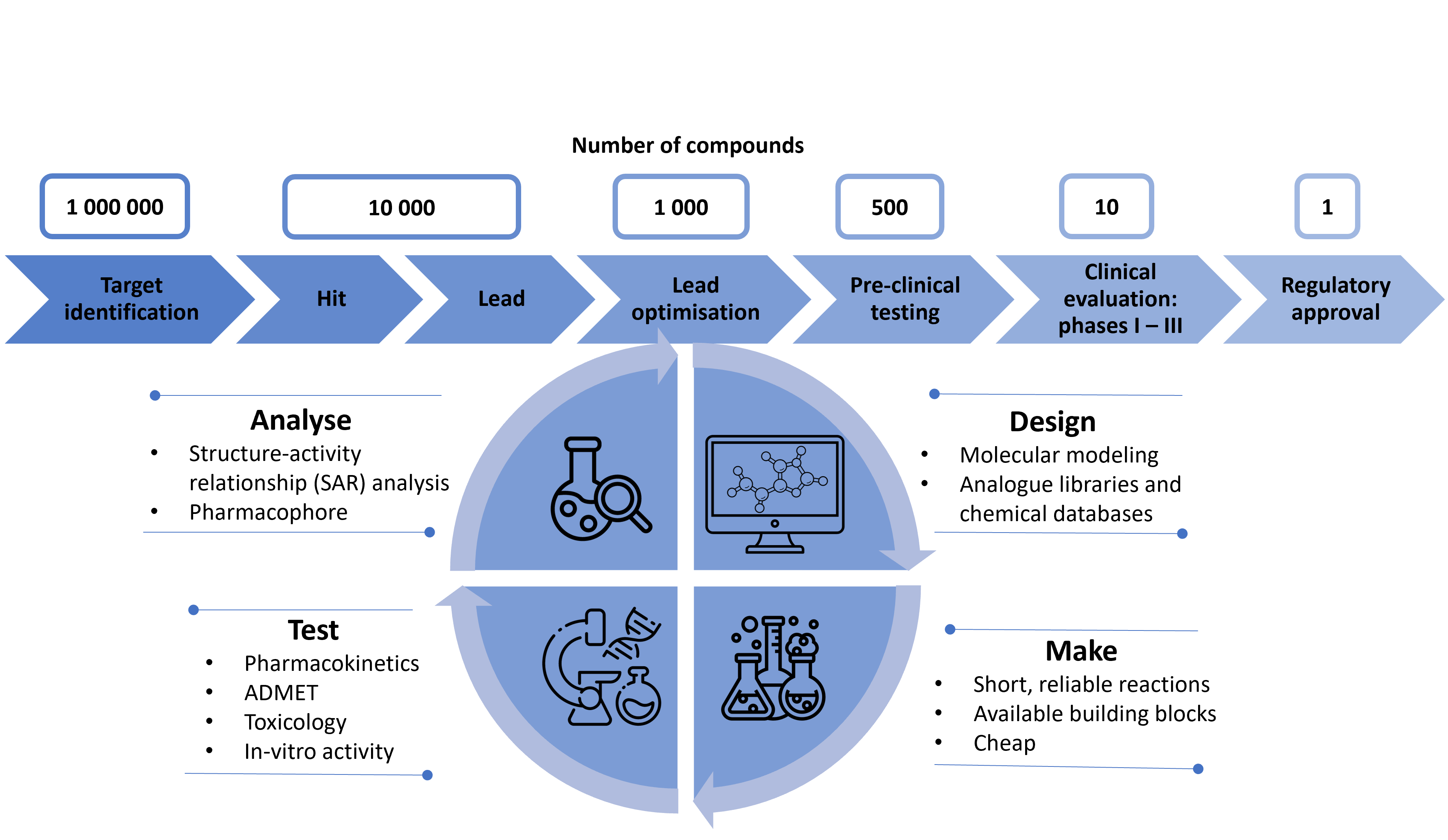
Have you ever wondered how new drugs are developed? It is actually quite a long and complex pathway and not only takes lots of time but also resources. The scheme below will help you understand the scale of that process.
Figure 1: A scheme showing drug discovery pipeline
As you can see, to get just one drug to the market, often thousands of structures are assessed to find the most effective and safe compound. That’s lots of information being generated in the whole process! The information can be stored in repositories and databases so that subsequent drug discovery process in a similar area is easier, faster and cheaper. Managing big data and using machine learning and computational chemistry methods is crucial in the lead optimisation step. By reviewing chemical databases and studying molecular docking simulations, we can save much time and resources to synthesise only those compounds that are the most promising based on in-silico methods. Even after synthesising the optimised ‘leads’, we still get lots of data from tests and analyses. By using appropriate cheminformatic tools and software, we can use the results to drive forward this iterative process of lead optimisation.
Ten Vs of big data for drug discovery
Hopefully, now you have an idea of what the process of drug discovery looks like. To understand why managing data in medicinal chemistry is so challenging, let’s have a closer look at the “Ten Vs” of big data Zhao et al. 2020:
Volume: size of data.
Velocity: speed of new data generation.
Variety: various formats of data.
Veracity: quality of data.
Validity: authenticity of data.
Vocabulary: terminology of data.
Venue: platform of data generation.
Visualization: view of data.
Volatility: duration of data usefulness.
Value: the potential of data usefulness to reduce the cost of drug discovery and development.
Medicinal chemists must keep in mind those features while both searching for data and publishing it. In this way we can focus on the main aspects of data management, try to improve the availability and normalisation of data, and be aware of limitations of repositories and inconsistencies in data quality.
FAIR MedChem
As you saw before, there are many properties that the compounds can be searched by, such as molecular weight, H-bond donors or acceptors, polarity, etc. Sometimes there is a need to assess other molecular properties but it might happen that not all the information is included in the database. This is one of the reasons why new repositories are being developed – they are more specific and gather particular properties of interest. How much easier the life of scientists could be if the relevant data was publicly available, well-ordered and contained the needed metadata? By submitting the data with the necessary information to the repository is a good way to make the data FAIR. This will make it:
Findable, as the data will be given specific identifiers
Accessible, as the data will be available online, open and free where possible
Interoperable, as the repository will often enforce the use of formalised, consistent language
Reusable, as the data will be released under a license with detailed provenance
In the repositories
Chemical and pharmacological databases
Currently, there are lots of publicly available databases storing information about hit molecules, chemical structures and properties, drug targets, pharmaceuticals… In the recent paper Zhao et al. 2020 the authors collated relevant databases to advance computer-aided drug discovery and divided them into several main groups:
Chemical collections
Drug / drug-like compounds
Drug targets, including genomics and proteomics data
Biological data from assay screening, metabolism, and efficacy studies
Drug liabilities and toxicities
Clinical databases
Below you will find the databases listed under corresponding categories, all taken from Zhao et al. 2020 paper.
There are many more databases available, and many are still being developed. They are often quite specific and contain certain types of compounds (eg. PROTACs or are aimed at a particular disease. If the listed databases are not specific enough for your research, you can try luck by searching smaller, more specific databases.
Contains chemical molecule (mostly small molecule) information, including chemical structures, identifiers, chemical and physical properties, biological activities, safety and toxicity data
97 million compounds, 236 million substances, 268 million bioactivities
Freely accessible database that currently holds 21 million virtual products originating from a small library of building blocks and collection of robust organic reactions
Manually curated database of bioactive molecules with drug-like properties; brings together chemical, bioactivity, and genomic data to aid translation of genomic information into effective new drugs.
>1.9 million compounds, 1.1 million pieces of assay information
Integration of a database and a data-mining system for network pharmacology analysis of all respects of traditional Chinese medicine, including herbs, herbal ingredients, targets, related diseases, adverse effect, and toxicity
Contains natural compounds, including information about corresponding 2D structures, physicochemical properties, predicted toxicity class and potential vendors
Screening library of 12 000 molecules assembled by combining three databases (Clarivate Integrity, GVK Excelra GoStar, and Citeline Pharmaprojects) to facilitate drug repurposing
Contains approved/marketed drugs with regulatory details, chemical structures (2D and 3D), dosage, biological targets, physicochemical properties, external identifiers, adverse effects, and PK data
Public, web-accessible database of measured binding affinities, focusing chiefly on interactions of proteins considered to be candidate drug targets with ligands that are small, drug-like molecules
1 756 093 binding data, for 7371 protein targets and 780 240 small molecules
Provides essential linkage between energetic and structural information of biomolecular complexes, which is helpful for various computational and statistical studies on molecular recognition in biological systems
The Biological General Repository for Interaction Datasets is an open-access database on protein, genetic, and chemical interactions for humans and all major model organisms
1 753 686 protein and genetic interactions, 28 093 chemical associations and 874 796 posttranslational modifications from major model organisms
Contains data from GPCRs, including crystal structures, sequence alignments, and receptor mutations; can be visualized in interactive diagrams; provides online analysis tools
GPCR-Ligand Association (GLASS) database is a manually curated repository for experimentally validated GPCR–ligand interactions; along with relevant GPCR and chemical information, GPCRligand association data are extracted and integrated into GLASS from literature and public databases
~ 277 651 unique ligands and 3048 GPCRs
Biological data from assay screening, metabolism, and efficacy studies
Therapeutic Target Database (TTD) is a database providing information about known and explored therapeutic protein and nucleic acid targets, targeted disease, pathway information and corresponding drugs directed at each of these targets
Database and knowledge inference system that integrates multiple bioactivity data sets to provide researchers with novel capabilities for mining and exploration of available structure-activity relationships (SAR) throughout chemical biology space.
Maintained by the National Cancer Institute; contains small-molecule information such as names, biological activities, and structures; a useful resource for researchers working in cancer/AIDS fields
Provides information on the plant-derived natural compounds, including structure, properties (physical, elemental, and topological), cancer type, cell lines, inhibitory values (IC50, ED50, EC50, GI50), molecular targets, commercial suppliers, and drug-likeness of compounds
Contains physiological parameter values for humans from early childhood through senescence; intended to be used in physiologically based (PB) PK modelling; also contains similar data for animals (primarily rodents)
Includes computational toxicology information about compounds, including HTS, chemical exposure, sustainable chemistry (chemical structures and physicochemical properties) and virtual tissue data
Contains drugs with potential to cause drug-induced liver injury in humans; established using FDA-approved prescription drug labels; liver-toxicity-knowledge-base-ltkb/ltkb-benchmark-dataset
Comparative Toxicogenomics Database (CTD) is a premier public resource for literature-based, manually curated associations between chemicals, gene products, phenotypes, diseases, and environmental exposures
Publicly available relational database that contains all information (protocol and result data elements) about every study registered in ClinicalTrials.gov. Content is downloaded from ClinicalTrials.gov daily and loaded into AACT
~ 324 429 research studies in all 50 US states and 209 countries
Contains detailed information on the nature of biomarkers, populations and subjects where measured, samples analyzed, methods used for biomarker analyses, concentrations in biospecimens, correlations with external exposure measurements, and biological reproducibility over time
A pharmacogenomics knowledge resource that encompasses clinical information about drug molecules
733 drugs with their clinical information
Data management in chemistry using Galaxy
Chemical file formats
Depending on the type of analysis you are going to perform, you will work with different file formats. Below are the most important data types, commonly used in cheminformatics, that you will likely see in Galaxy:
SDF (.sd, .sdf) - stands for Structure-Data Format, and SDF files wrap the molfile (MDL Molfile) format. Stores information about the chemical structure and associated data of compounds in plain text. Files in SDF format can encode single or multiple molecules that are then delimited by lines consisting of four dollar signs. SDF files are formatted ASCII files that store information about the positions of the individual atoms (either in 2D or 3D space) that make up the molecule. The data on connectivity and hybridization state are also encoded, although their use is less frequent and often inconsistent.
MOL (.mol) - an MDL Molfile for holding information about the atoms, bonds, connectivity and coordinates of a molecule.
MOL2 (.mol2) - a Tripos Mol2 file is a complete, portable representation of a SYBYL molecule. It is an ASCII file which contains all the information needed to
reconstruct a SYBYL molecule
CML (.cml) - Chemical Markup Language (ChemML or CML) is an approach to managing molecular information using tools such as XML and Java. It supports a wide range of chemical concepts including molecules, reactions, spectra and analytical data, computational chemistry, chemical crystallography and materials
InChI (IUPAC International Chemical Identifier) - a textual identifier for chemical substances, designed to provide a standard way to encode molecular information. The identifiers describe chemical substances in terms of layers of information — the atoms and their bond connectivity, tautomeric information, isotope information, stereochemistry, and electronic charge information (Heller et al. 2015)
SMILES (.smi) - the simplified molecular-input line-entry system (SMILES) is a specification in the form of a line notation for describing the structure of chemical species using short ASCII strings. A linear text format which can describe the connectivity and chirality of a molecule (Weininger 1988)
PDB - the Protein Data Bank (PDB) file format is a textual file format describing the three-dimensional structures of molecules held in the Protein Data Bank, now succeeded by the mmCIF format. It contains a description and annotation of protein and nucleic acid structures including atomic coordinates, secondary structure assignments, as well as atomic connectivity. In addition, experimental metadata is stored. (Berman 2007)
GRO (.gro) - a plain text file storing spatial coordinates and velocities (if available) of atoms during a molecular dynamics simulation, utilised by GROMACS
To visualise the structures held by the files with the positions of the individual atoms, you can use (NGL Viewer)[https://nglviewer.org/ngl/].
If you plan to work with molecular dynamics simulations, there are also some MD trajectory file formats that you might want to get familiar with. In Galaxy, you can convert between xtc, trr, dcd and netcdf files using a tool called MDTraj file converter ( Galaxy version 1.9.6+galaxy0).
Galaxy tools
In Galaxy Chemical Toolbox there are dozens of tools that can be used for various analyses. Below we will only show a few, mostly related to data import, format conversion and some functions linked to what was discussed previously.
Let’s start with importing publicly available data. Protein Data Bank stores thousands of three-dimensional structural data of proteins nucleic acids and other biological molecules.
Figure 3: How to get accession code of the resulting structures.
Switch to Galaxy
Get PDB file ( Galaxy version 0.1.0) with the following parameters:
“PDB accession code”: 2BK3
This is often the first step of docking studies that you can learn about in other tutorials, for example in Protein-ligand docking tutorial.
You can import any molecule to Galaxy using SMILES notation. Below we show an example with benzenesulfonyl chloride and ethylamine.
Hands On: Import SMILES
Copy SMILES of your molecule(s) of interest. In this example, we use benzenesulfonyl chloride (SMILES: C1=CC=C(C=C1)S(=O)(=O)Cl) and ethylamine (SMILES: CCN).
Click on the galaxy-upload Upload data button in the tools panel on the left-hand side.
Click on the galaxy-wf-edit Paste/Fetch data button twice. Two boxes will appear.
In the first box, under “Name” section, enter Benzenesulfonyl chloride and under “Type”: smi
Below paste the SMILES of benzenesulfonyl chloride: C1=CC=C(C=C1)S(=O)(=O)Cl
In the second box, under “Name” section, enter Ethylamine and under “Type”: smi
Below paste the SMILES of ethylamine: CCN
Click the “Start” button, then close the dialogue box. Your files are being added to your history!
As you learned from the previous section, there are many different formats used in computational chemistry. Of course, Galaxy allows you to interconvert between them - just have a look below!
Hands On: Convert the file format
Compound conversion ( Galaxy version 3.1.1+galaxy0) with the following parameters:
param-files“Molecular input file”: click on param-filesMultiple datasets icon and choose both Benzenesulfonyl chloride and Ethylamine
“Output format” - you have many options to choose from! Not only the formats but also the associated parameters. Just scroll the list and pick the one that is relevant to your downstream analysis. Here we use MDL MOL format (sdf, mol)
Rename galaxy-pencil the corresponding files Benzenesulfonyl chloride SDF and Ethylamine SDF. Before renaming the files, make sure to check either the starting dataset numbers or the atoms in the file in order not to confuse the two files!
We intentionally chose to work on benzenesulfonyl chloride and ethylamine. Do you know why? Well, if you think about the synthesis of sulfonamides, that’s exactly what you need - a sulfonyl chloride and an amine. So… let’s do some computational synthesis!
Hands On: Run the reaction
Reaction maker ( Galaxy version 1.1.4+galaxy0) with the following parameters:
Above we converted the .smi file into .sdf to show how toolCompound conversion tool works. However, it is worth pointing out that toolReaction maker works also with SMILES files and the conversion happens automatically so that your .smi dataset is imported as .sdf input.
toolReaction maker works with more reaction types, such as:
The output is the .txt log file and the SDF file with the product molecule. If you have a look at that file, you’ll see that there is just a list of atoms with their corresponding coordinates. To draw the resulting structure, we can use another tool.
Hands On: Visualisation of compounds
Visualisation ( Galaxy version 3.1.1+galaxy0) with the following parameters:
param-file“Molecular input file”: SDF output for Reaction maker
“Property to display under the molecule” - there are many properties that you can choose from! Please note that sometimes if not enough information is provided, then the property might not be displayed. Let’s choose a basic but useful property: Molecular weight
“Format of the resulting picture”: SVG
Since we spoke about the drug-likeness in the previous section, let’s see how it works in practice, on the example of our “synthesised” molecule.
Hands On: Estimate the drug-likeness
Drug-likeness ( Galaxy version 2021.03.4+galaxy0) with the following parameters:
param-file“Molecule data in SDF or SMILES format”: SDF output for Reaction maker
“Method” - two possible methods to weight the features are available - one based on max. weight (QEDw,max), and the other on mean weight (QEDw,mo). There is also an option to leave features unweighted (QEDw,u) and we’ll choose this one: unweighted (QEDw,u)
“Include the descriptor names as header”: param-toggleYes
The eight properties used are molecular weight (MW), octanol–water partition coefficient (ALOGP), number of hydrogen bond donors (HBDs), number of hydrogen bond acceptors (HBAs), molecular polar surface area (PSA), number of rotatable bonds (ROTBs), number of aromatic rings (AROMs) and number of structural alerts (ALERTS).
In medicinal chemistry, we often base the new structures on scaffolds of existing drugs. Therefore, it is quite useful to be able to find similar structures - below is an example of how to do it in Galaxy.
Hands On: Search ChEMBL database for similar compounds
Search ChEMBL database ( Galaxy version 0.10.1+galaxy4) with the following parameters:
“SMILES input type”: File
param-file“Input file”: Benzenesulfonyl chloride
“Search type”: Substructure
Rename galaxy-pencil the output file Benzenesulfonyl chloride substructures
Even though the input format of the above tool is SMILES, it can automatically interconvert between .sdf and .smi formats, so you don’t have to use toolCompound conversion tool beforehand.
toolSearch ChEMBL database tool allows for searching the ChEMBL database for compounds which resemble a SMILES string. Two search options are possible:
similarity (searches for compounds which are similar to the input within a specified Tanimoto cutoff)
substructure (searches for compounds which contain the input substructure)
Results can be filtered for compounds which are
approved drugs
biotherapeutic
natural products
fulfil all of the Lipinski rule of five criteria
Let’s repeat this step, but with Lipinski’s Rule of Five which you learned about in the previous section of this tutorial!
Hands On: Search ChEMBL database for similar compounds using the Lipinski rule of five
Click the galaxy-refreshRepeat button on the previous dataset.
You will be redirected to Search ChEMBL database ( Galaxy version 0.10.1+galaxy4). Leave all the parameters as before, except for one:
“Filter for Lipinski’s Rule of Five”: param-toggleYes
Rename galaxy-pencil the output file Benzenesulfonyl chloride Lipinski substructures
Question
How many molecules have been filtered out?
If you click on the dataset, you will see a short summary of how many molecules have been found.
When we didn’t apply any filters, the tool found 45 molecules. After applying Lipinski’s Rule of Five filter, 36 molecules satisfied the rules.
Figure 4: Preview showing the number of molecules found in the ChEMBL database, before and after applying Lipinski's Rule of Five filter
Galaxy workflow summary
Those are just a few tools, but they offer many parameters that you can tune depending on your analysis. Additionally, we provide you with an example history and the dedicated workflow of this small tutorial. You are more than welcome to explore other tools in the Galaxy Chemical Toolbox though!
Data-driven medicinal chemistry - yesterday, today and tomorrow
Why do we need big data in med-chem?
In the article by Lusher et al. 2014 the role of the medicinal chemist is to make decisions about which of the infinite possibilities of new compounds should be made next. As the amount and variety of data on which to base these decisions grows, so too must the data analysis skills of the medicinal chemist. The authors claim that modern medicinal chemists should be able to recognise sources of relevant information, prepare raw data, use statistical tools, extract meaningful information, interpret results, recognise potential problems and make visualisations to communicate their findings to improve the quality of compounds being produced in research. According to the authors, this situation will require improved education and increased access to data and information management tools. This is where Galaxy comes in, offering not only a platform for data analysis with a plethora of the most commonly used tools in the field but also educational materials allowing anyone to learn and excel in analysis.
The privilege of applying computational methods in medicinal chemistry pipelines allows to focus on the most promising compounds and to remove unsuitable ones before the stage of chemical synthesis. Not only is this approach more efficient but also more sustainable and can reduce the costs of the synthetic stage of drug discovery (Brown et al. 2018).
Another important role that big data plays in drug discovery is target identification (and validation) which might be currently one the biggest challenges in medicinal chemistry. By introducing genomic data in drug discovery, we can produce more specific and effective medicines. Understanding the underlying causes of the disease and the biological targets are crucial in designing new drugs and hence should be inherently linked with this process, where possible.
Data challenges
It is important to understand the limitations of the databases we use since if data is not well understood, the processing and analysis drawn may ultimately be flawed, following the ‘garbage in, garbage out’ principle. Here are some challenges mentioned in Brown et al. 2018 that you can explore more in-depth by referring to that article.
Errors
Reproducibility
Standardisation
Formatting of data
Information held in silos
Loss of contextual information
Limited and biased data
Machine learning in med-chem
Given the huge number of molecules in the various databases, applying machine learning in cheminformatics and building predictive models of the physiochemical properties of molecules is more and more popular. It helps to predict absorption, distribution, metabolism, excretion, toxicity (ADMET), likelihood of interaction with the drug targets as well as off-target effects. It can be useful in elucidating complex protein-protein or drug-drug interaction networks, in understanding structure-activity relationships (SARs) and when combined with data from the ‘omics’ revolution. This approach also helps to identify bioisosteres and introduce the idea of scaffold hopping in molecular design (Brown et al. 2018).
Knowledge sharing
It is not only about gathering and managing the data but also building a culture of knowledge sharing. We work in big, international teams with well-established methods and well-equipped labs. There are so many opportunities for knowledge capture and exchange! The challenge emerges here though – how to efficiently connect and help each other? This is the question that many pharmaceutical companies try to answer, such as Merck & Co. who describe their approach to knowledge management and report on the multiple enduring and complementary teams and initiatives to capture and share knowledge (Beshore et al. 2022).
You've Finished the Tutorial
Please also consider filling out the Feedback Form as well!
Key points
The power of big data might be useful to shape innovations of the future in the pharmaceutical field.
There are many publicly available databases used for drug discovery and development and they might look at the same medicines from different angles and classify them based on various factors.
Galaxy provides tools and a platform for medicinal chemistry analyses.
Frequently Asked Questions
Have questions about this tutorial? Have a look at the available FAQ pages and support channels
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
References
Weininger, D., 1988 SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. Journal of Chemical Information and Computer Sciences 28: 31–36. 10.1021/ci00057a005
Ghose, A. K., V. N. Viswanadhan, and J. J. Wendoloski, 1999 A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. Journal of Combinatorial Chemistry 1: 55–68. 10.1021/cc9800071
Egan, W. J., K. M. Merz, and J. J. Baldwin, 2000 Prediction of Drug Absorption Using Multivariate Statistics. Journal of Medicinal Chemistry 43: 3867–3877. 10.1021/jm000292e
Lipinski, C. A., F. Lombardo, B. W. Dominy, and P. J. Feeney, 2001 Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings 1PII of original article: S0169-409X(96)00423-1. The article was originally published in Advanced Drug Delivery Reviews 23 (1997) 3–25. 1. Advanced Drug Delivery Reviews 46: 3–26. 10.1016/s0169-409x(00)00129-0
Muegge, I., S. L. Heald, and D. Brittelli, 2001 Simple Selection Criteria for Drug-like Chemical Matter. Journal of Medicinal Chemistry 44: 1841–1846. 10.1021/jm015507e
Veber, D. F., S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward et al., 2002 Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. Journal of Medicinal Chemistry 45: 2615–2623. 10.1021/jm020017n
Martin, Y. C., 2005 A Bioavailability Score. Journal of Medicinal Chemistry 48: 3164–3170. 10.1021/jm0492002
Berman, H. M., 2007 The Protein Data Bank: a historical perspective. Acta Crystallographica Section A Foundations of Crystallography 64: 88–95. 10.1107/s0108767307035623
Lusher, S. J., R. McGuire, R. C. van Schaik, C. D. Nicholson, and J. de Vlieg, 2014 Data-driven medicinal chemistry in the era of big data. Drug Discovery Today 19: 859–868. 10.1016/j.drudis.2013.12.004
O′Hagan, S., N. Swainston, J. Handl, and D. B. Kell, 2014 A ‘rule of 0.5’ for the metabolite-likeness of approved pharmaceutical drugs. Metabolomics 11: 323–339. 10.1007/s11306-014-0733-z
Heller, S. R., A. McNaught, I. Pletnev, S. Stein, and D. Tchekhovskoi, 2015 InChI, the IUPAC International Chemical Identifier. Journal of Cheminformatics 7: 10.1186/s13321-015-0068-4
Daina, A., O. Michielin, and V. Zoete, 2017 SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports 7: 10.1038/srep42717
Brown, N., J. Cambruzzi, P. J. Cox, M. Davies, J. Dunbar et al., 2018 Big Data in Drug Discovery, pp. 277–356 inProgress in Medicinal Chemistry, Elsevier. 10.1016/bs.pmch.2017.12.003
Mullard, A., 2018 Re-assessing the rule of 5, two decades on. Nature Reviews Drug Discovery 17: 777–777. 10.1038/nrd.2018.197
Zhao, L., H. L. Ciallella, L. M. Aleksunes, and H. Zhu, 2020 Advancing computer-aided drug discovery (CADD) by big data and data-driven machine learning modeling. Drug Discovery Today 25: 1624–1638. 10.1016/j.drudis.2020.07.005
Beshore, D. C., A. M. Haidle, A. Arasappan, Y.-H. Lim, I. Raheem et al., 2022 Building a Culture of Medicinal Chemistry Knowledge Sharing. Journal of Medicinal Chemistry 65: 3776–3785. 10.1021/acs.jmedchem.1c02144
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.
Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{computational-chemistry-med-chem-data,
author = "Julia Jakiela and Katarzyna Kamieniecka and Krzysztof Poterlowicz",
title = "Data management in Medicinal Chemistry (Galaxy Training Materials)",
year = "",
month = "",
day = "",
url = "\url{https://training.galaxyproject.org/training-material/topics/computational-chemistry/tutorials/med-chem-data/tutorial.html}",
note = "[Online; accessed TODAY]"
}
@article{Hiltemann_2023,
doi = {10.1371/journal.pcbi.1010752},
url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752},
year = 2023,
month = {jan},
publisher = {Public Library of Science ({PLoS})},
volume = {19},
number = {1},
pages = {e1010752},
author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and},
editor = {Francis Ouellette},
title = {Galaxy Training: A powerful framework for teaching!},
journal = {PLoS Comput Biol}
}
Funding
These individuals or organisations provided funding support for the development of this resource
Questions:
 Open image in new tab
Open image in new tabOpen image in new tab
Open image in new tab
Open image in new tab

